-
A bugfix was made for Antechamber-1.27
-
Antechamber-1.27 has been released on the Antechamber website on August 17.
Here is the improvement over 1.26
- Modified mopcrt.c for mopac to do optimizations for excited states. A typical command is like: $ACHOME/exe/antechamber -fi mol2 -fo mol2 -i test.mol2 -o test_bcc.mol2 -c bcc -nc NET_CHAEGE -m MULTIPLICITY -j 4 -df 0
- Fixed two bugs about memory allocation in bondtype.C
- Improved database.c and it is now more robust
Download the latest version of Antechamber package Version 1.27. .
Antechamber-1.26 has been released on the Antechamber website on June 29.
The following is the new features of 1.26:
- Modified the charge.c function and the parameters of calculating the Gasteiger charges. The parameters of a,b,c,d are from those of Gasteiger et al. (J. Gasteiger, M. Marsili, Tetrahedron, 36, 3219-3228 (1980)) except those for phosphate, which are borrowed from sybyl7.2. The charges calculated by the antechamber program may slightly different from those by sybyl due to a more stringent converge criterion is applied. Unlike sybyl, which needs the users to assign atomic formal charges before the charge calculation, the antechamber.c program could assign formal charges automatically.
- Added a new function of spaceline() to judge if a line is a space line (only has spaces or control characters) or not. We found that strlen(line) == 1 fails for files generated by Windows (there is cntr-M at the end of each line either visable or invisable)
- Revised prep.c to read in the atom connectivity information from a prepi file and no atomic connectivity is judged for the prepi input. Now antechamber and parmchk wrok properly even when the input structures are not good enough.
- Created an atom type description file for Tripos atom types (ATOMTYPE_SYBYL.DEF) and revised the atomtype.c program to recognize sybyl atom types. The wmol2() funcition in mol2.c was also modified accordingly.
- Improved database.c greatly. Now database.c can handle almost any multiple-record database file only if it is well organized (some symbols to separate each record, such as "$$$$" of sdf, "@MOLECULE" of multiple-mol2 files). It can even properly handle a file like all_amino94.in in $ACHOME/dat/leap/prep. A sample input file (sample.def) is provided in the $ACHOME/test/database.
- Added a new atom type, cz for the sp2 carbon in a guadinium group. It was found by Cristophe Guilbert at UCSF that the original gaff.dat parameter set breaks the planar conformation of 2-amino-1H-benzo[d]imidazol-3-ium. We studied a set of similar molecules and found that only 2-amino-1H-benzo[d]imidazol-3-ium (the first molecule) caused trouble. We solved this problem by simple applying a larger force constant of improper dihedral angles involved by cz (X -X -cz-X 10.5 180. 2.). All the other parameters involved by cz are borrowed from those of c2. We think it may not a good idea to introduce atom types for five-memberred rings because (1) many atom types (sp2 C, sp3 C, sp2 N etc) are added; (2) the current parameter set works fine for most of molecules that have five-memberred rings.
Download the latest version of Antechamber package Version 1.26. .
How to generate proper topology files (run QM to get RESP charges, assign proper atom types) while keeping the coordinates from the crystal structures? Here is some tips.
There is a glitch in ring.c found by Eric Pettersen from UCSF. For a certain types of computer systems, there may have a memory core dump problem for some kinds of molecules. The following modification is made on the ring.c in Antechamber 1.25:
68,69c68
< if(ringnum == 0)
< return;
---
>
The modified version looks like:
64 RING *ringbak;
65 int ringbaknum = 0;
66 int tmpint;
67 int index;
68 if(ringnum == 0)
69 return;
70 ringbak = (RING *) malloc(sizeof(RING) * ringnum );
71 if (ringbak == NULL) {
72 fprintf(stderr, "memory allocation error
for *ringbak\n" );
73 exit(0);
Some glitches were fixed for generating Tripos sybyl atom types. Download the latest version of Antechamber package Version 1.25. .
A new program, charmmgen, which writes gaff parameters in charmm format, is added into the Antechamber package. A bug was fixed for parmchk.c and gaff.dat is updated to remove some glitches. Download the latest version of Antechamber package Version 1.24. .
A bug was fixed for parmchk.c. Download the latest version of Antechamber package Version 1.23. .
Antechamber 1.2 has been released on Antechamber web page on 11/16/05. A new program, top2mol2, which reads in AMBER topology and restart or crd files to produce a mol2 file that contains
bond type information. It may be useful if one wants to quick check a minimized structure or the lastest MD structure in a rst file. Some bugfixes have been done on version 1.1.
Download the latest version of the Antechamber package Version 1.2. .
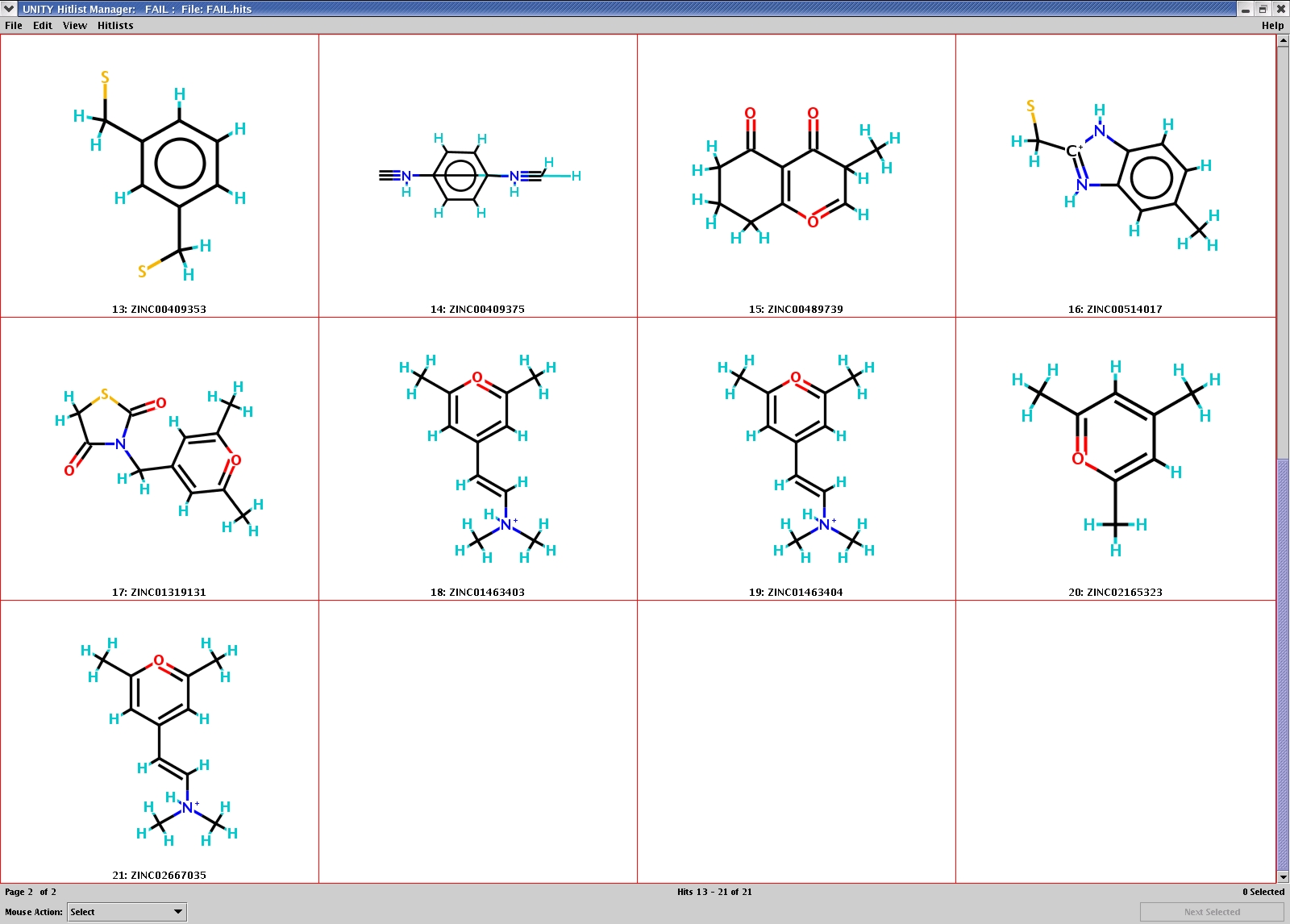
Antechamber, 1.2, has been extensively tested by more than 56,000 molecules in ZINC databases. You may download the fragment-like subset (multiple tautomeric and protonation states per chemical entity) in mol2 file and copy it to $ACHOME/test/database and then run "Run.database" to test the package on your computer (the name of the multiple mol2 file is xpaa.mol2). There were 21 out of 56,000 molecules that cause failure (successful rate was estimated to be 99.96%). Here are the 2D structures ( Set 1 and Set 2 ) of the failed molecules. Once could see that those structures are quite weird. The total processing time was about 85 minutes running on a P4 1.8 MHz Linux machine.
Antechamber 1.1 has been released on Antechamber web page on 11/10/05. Many features have been added in this version and here is the details .
Download the latest version of the Antechamber package Version 1.1.
We have decided to make antechamber a free software with a license under the GNU General Public Library
We have tested the package throughly with 7722 molecules in CMC (Comprehensive Medical Chemistry) database. Two commands were performed for all the molecules, which are:
/home/jwang/antechamber/antechamber-1.0/bin/antechamber -fi mol2 -fo prepi -i $mol.mol2 -o $mol.prep1 -j 4
/home/jwang/antechamber/antechamber-1.0/bin/antechamber -fi mol2 -fo prepi -i $mol.mol2 -o $mol.prep1 -j 5
The processing time of running the two commands is about 0.3s per molecule. With a slightly improved version of antechamber in AMBER8, the bond types of 12 molecules could not be assigned correctly by running the first command (do both bond type and atom type prediciton). The successful rate is estimated to be 99.8 %. With the current version, all 7722 molecules can be successfully assigned bond types without error message. As to the second command (read in mol2 bond types, do bond type correction and atom type prediction), it always generates topology files without error message.
We have worked out several examples and tutorials to illustate how to use antechamber programs.
We have designed a trouble shooting page to help user solve the problem on antechamber.
The second generation of the general AMBER force field (GAFF) has been finished development. The paper describing GAFF has been published: J. Comput. Chem. 25, 1157-1174 (2004).
We have finished manscript writting for the Antechamber paper, which will be submitted to Journal of Chemical Information and Computer Science
For amber7 users, here is the sample file of mopac.sh for mopac7 and mopac8 and mopac508mn .
According to the definitions of h4 and h5 in ATOMTYPE_GFF.DEF of amber8, h4 and h5 are hydrogens attached to aromatic carbon with one or two eletron
withdrawing groups, respectively. Now we think this rule should be applied to all kinds of sp2 carbon, such as sp2 carbon in
formaldehyde and formamide. Thanks Dr. Astrid Maass of SCAI, Germany pointing out this. The current version has corrected this problem.
|
{kind=link}
{kind=link}