Simulating Crystals with the AMBER - System Setup¶
--- Jupyter notebook version ---
by Hai Nguyen (06/2016)
Original version: http://ambermd.org/tutorials/advanced/tutorial13/XtalTutor1.html
from IPython.display import IFrame
IFrame(src="view.html", width=550, height=550)
Requirements¶
You are advanced users
You successfully installed AmberTools 16 (or greater)
Files
- Download tar files and untar it (XtalUtilities.tar.gz): http://ambermd.org/tutorials/advanced/tutorial13/Tarball.html
- Download this notebook and copy it to XtalUtilities folder
- cd XtalUtilities
Install external library for protein viewer: nglview
$ amber.conda install nglview==0.5.1 -c ambermd
Open this notebook
$ amber.jupyter notebook crystal_simulation_setup.ipynb
How to run this notebook¶
click on each cell border, hit "Ctrl-Enter"
Need help? click "Help --> Notebook Help" at the top of this notebook
Notes: I keep the same title as the original tutorial
1. Constructing a lattice: You'll need some more tools for this!¶
Doc: http://ambermd.org/tutorials/advanced/tutorial13/Tarball.html
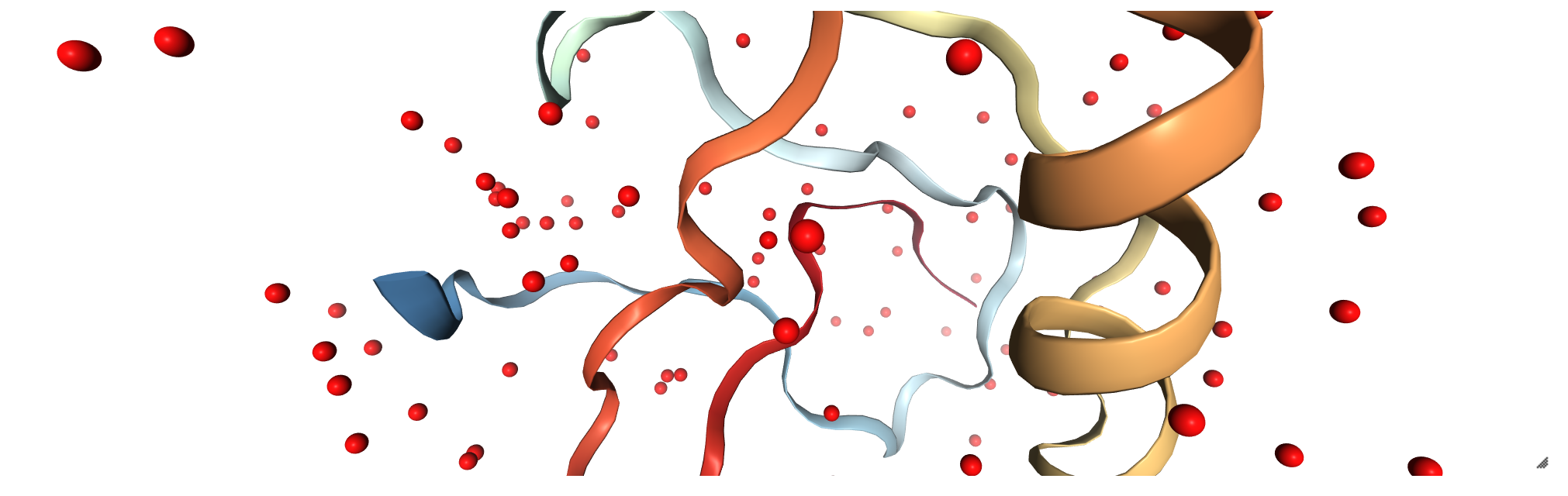
Viewing original pdb file¶
import pytraj as pt, nglview as nv
print(pt.__version__, nv.__version__)
traj = pt.load("c1AHO.pdb")
view = nv.show_pytraj(traj)
view
view.add_licorice('water and not hydrogen')
# use view._clear_repr() to clear all representations
You should expect to see
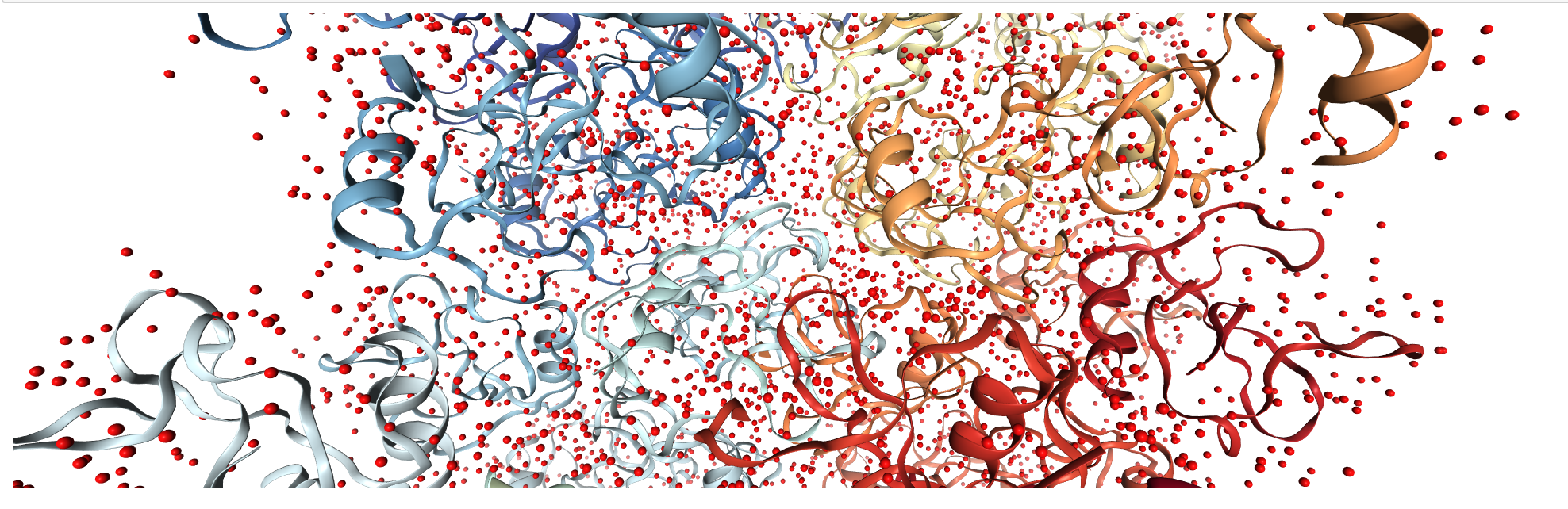
Constructing the unit cell: Where crystal simulations depart from ordinary MD¶
Doc: http://ambermd.org/tutorials/advanced/tutorial13/Construction.html
!${AMBERHOME}/bin/UnitCell -p c1AHO.pdb -o x1AHO.pdb
!${AMBERHOME}/bin/PropPDB -p x1AHO.pdb -o x8_1AHO.pdb -ix 2 -iy 2 -iz 2
traj2 = pt.load("x8_1AHO.pdb")
view2 = nv.show_pytraj(traj2)
view2
view2.add_licorice("water and not hydrogen")
You should expect to see
2. Solvating the unit cell: Filling in the gaps¶
Doc: http://ambermd.org/tutorials/advanced/tutorial13/Solvation.html
! ${AMBERHOME}/bin/AddToBox -c x1AHO.pdb -a Acetate.pdb -na 7 -o xa1AHO.pdb -P 4143 -RP 3.0 -RW 6.0 -G 0.2 -V 1
! ${AMBERHOME}/bin/AddToBox -c xa1AHO.pdb -a Ammonium.pdb -na 3 -o xm1AHO.pdb -P 4192 -RP 3.0 -RW 6.0 -G 0.2 -V 1
! ${AMBERHOME}/bin/AddToBox -c xm1AHO.pdb -a spce.pdb -na 179 -o solv1AHO.pdb -P 4192 -RP 3.0 -RW 3.0 -G 0.2 -V 1
3. Generating a topology: Working with what we have¶
Doc: http://ambermd.org/tutorials/advanced/tutorial13/Topology.html
%%file solvate.tleap
source leaprc.ff99SB_spce
loadAmberPrep Acetate.prp
loadAmberPrep Ammonium.prp
ammparms = loadAmberParams Ammonium.frcmod
x = loadPdb "solv1AHO.pdb"
bond x.11.SG x.62.SG
bond x.15.SG x.35.SG
bond x.21.SG x.45.SG
bond x.25.SG x.47.SG
bond x.204.SG x.255.SG
bond x.208.SG x.228.SG
bond x.214.SG x.238.SG
bond x.218.SG x.240.SG
bond x.397.SG x.448.SG
bond x.401.SG x.421.SG
bond x.407.SG x.431.SG
bond x.411.SG x.433.SG
bond x.590.SG x.641.SG
bond x.594.SG x.614.SG
bond x.600.SG x.624.SG
bond x.604.SG x.626.SG
set x box {45.900 40.700 30.100}
set default nocenter on
saveAmberParm x solv1AHO.top solv1AHO.crd
quit
! tleap -f solvate.tleap > solvate.out
! tail -20 solvate.out
! ${AMBERHOME}/bin/ChBox -c solv1AHO.crd -o solv1AHO.crd -X 45.9 -Y 40.7 -Z 30.1 -al 90.0 -bt 90.0 -gm 90.0
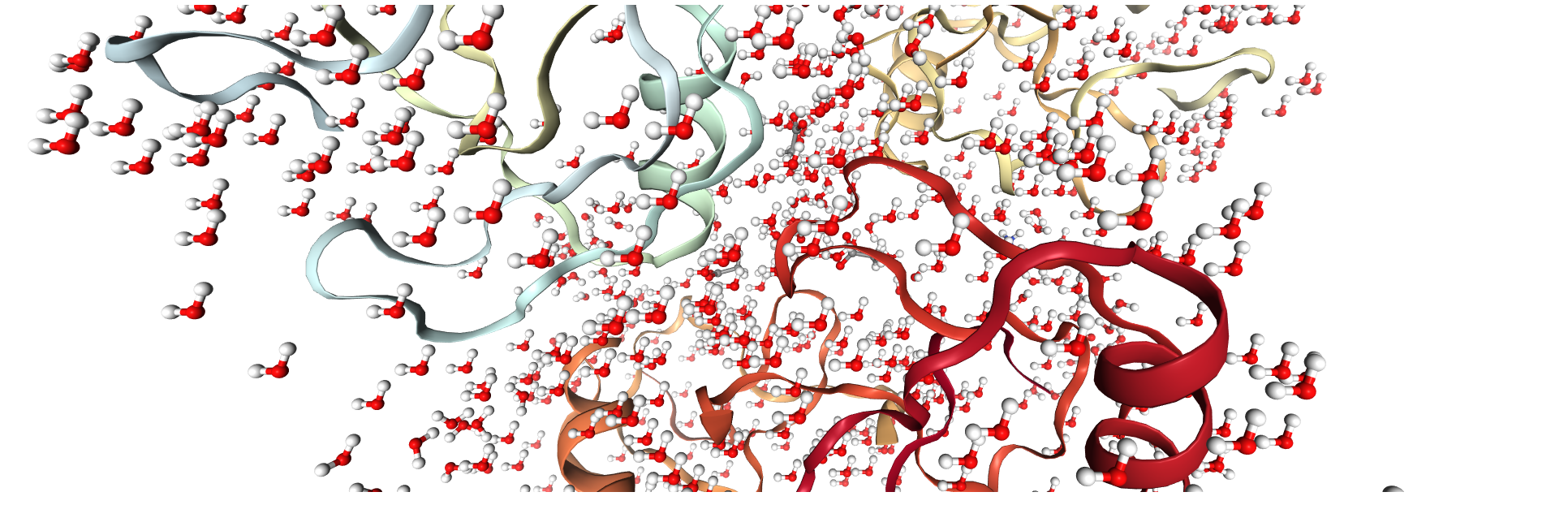
traj3 = pt.load("solv1AHO.crd", "solv1AHO.top")
view3 = nv.show_pytraj(traj3)
view3
view3.add_licorice('water and not hydrogen')
you should expect too see
# check the unitcells
traj3.unitcells