Copyright (C) Pengfei Li 2016
Tutorial for the LEaP Program
By Pengfei Li
Background Information
I. Terminology used in this tutorial
Before we begin the tutorial for the LEaP program, we should cover some
terminology that we will use in the tutorial.
In the classical force field, a molecule can be described as a collection of
many atoms of numerous types connected to each other, in which the bonded
interaction between atoms is represented by bond, angle and dihedral terms
while the nonbonded interaction is treated as sum of electrostatic and var
der Waals (VDW) interactions.
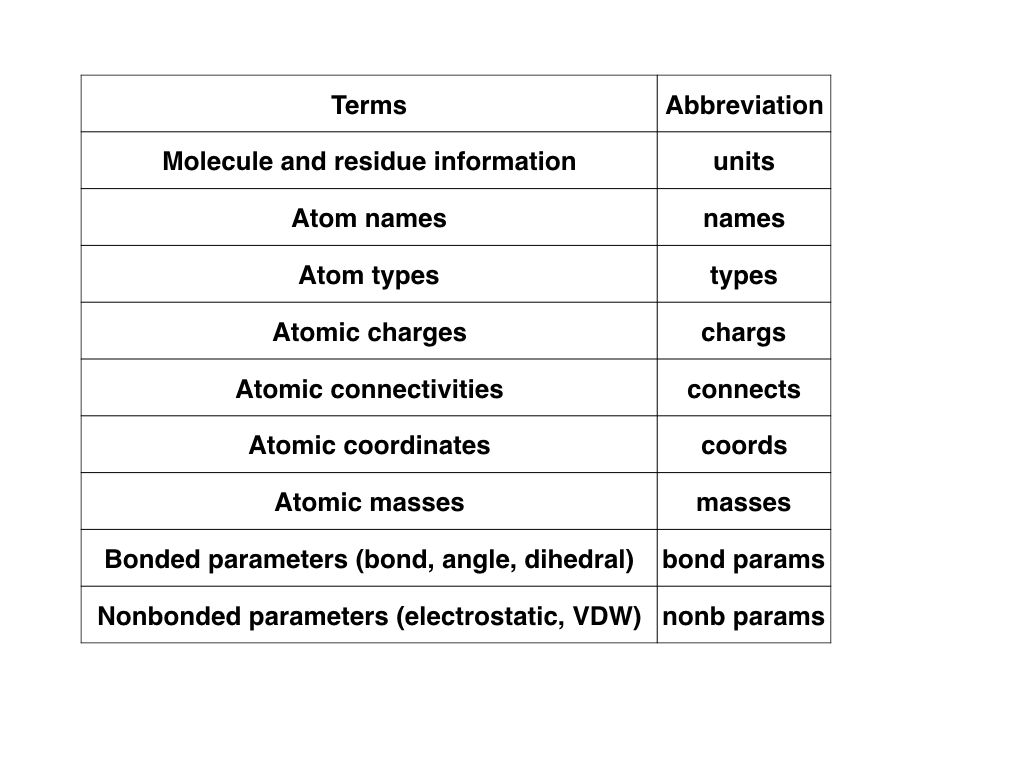
The following figure showed the minimum information (additional information
may be needed under some specific situations) required to define a molecule
in a classical force field.
The corresponding numbers for each term are used in the other figures of
this tutorial.
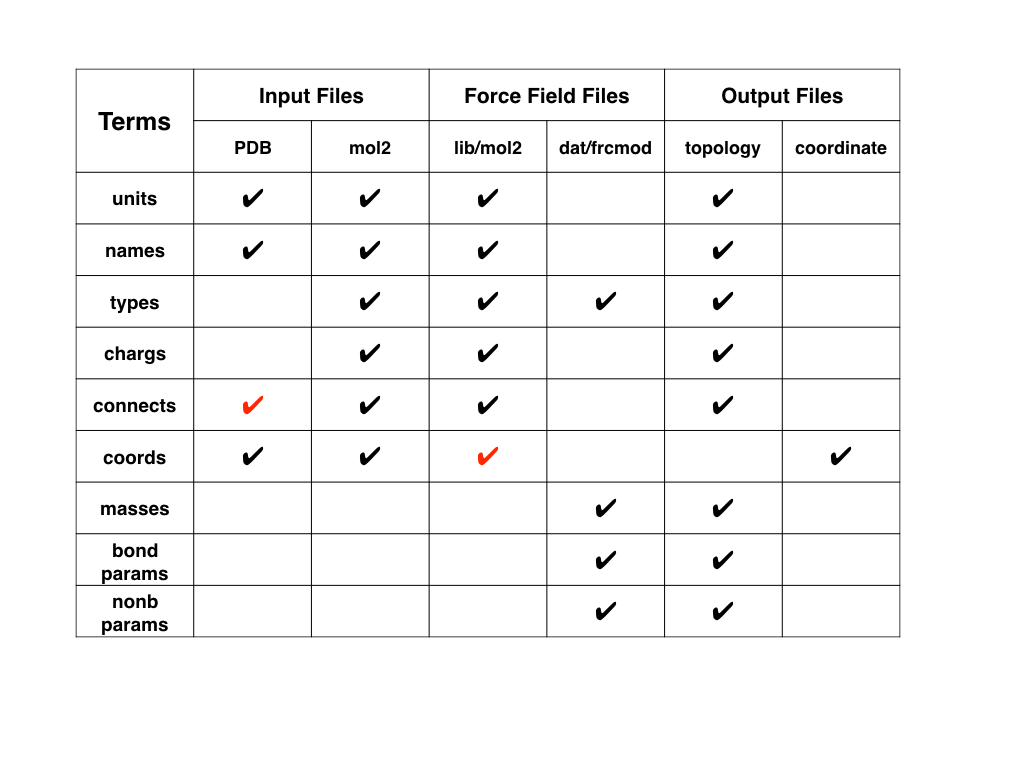
II. Different files used in AMBER
A lot of new learners are confused about how to use AMBER, this especially
applies to how the LEaP program works and the differences between various
files (such as pdb, mol2, lib, prep, dat, frcmod).
Each of these files contains part of the information needed for performing a
minimization or molecular dynamics simulation based on classical force field,
and LEaP can generate AMBER topology file (with suffix as "prmtop", "parm7",
"top") and coordinate file (or suffix as "inpcrd", "restrt", "rst7", "crd")
based on them.
Note: a basic information flow about the entire AMBER software package has
been shown in section 1.1 of the
AMBER 2017 Manual,
which could be helpful for users to have a big picture of the general modeling
process.
Here is a figure illustrating the information that these different files have.
The black cross in the figure means this part information is contained and used
for the file generation process of LEaP.
The red cross in the figure indicates this part of information may be contained
in the original file but usually not used for the file generation process of
LEaP.
For more detailed information about the contents of each file and their
formats, please see the
AMBER file formats webpage.
And in section 14.1 of the
AMBER 2017 Manual
there is a detailed information of the Amber parameter file.
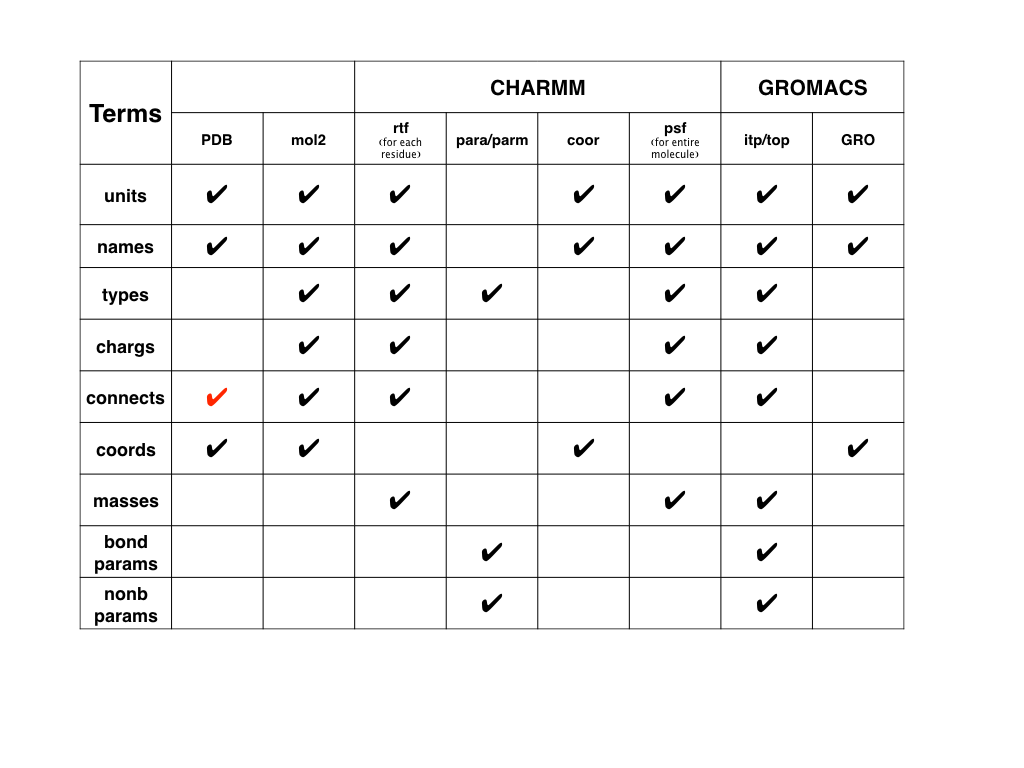
Here we also have a figure for modeling files in CHARMM and GROMACS.
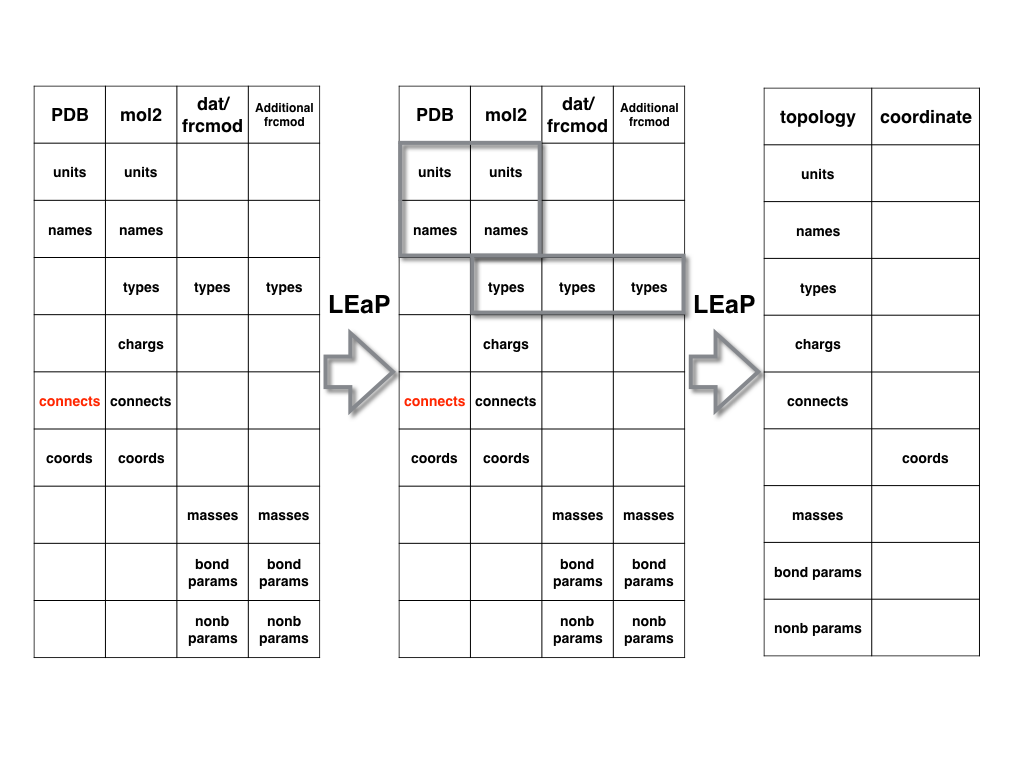
How Does LEaP Work?
The relationship between each file in above figure and LEaP can be described in
the following manner: the files are akin to independent boards while LEaP is
like a carpenter nails/merges them together based on their overlapping parts,
as shown below:
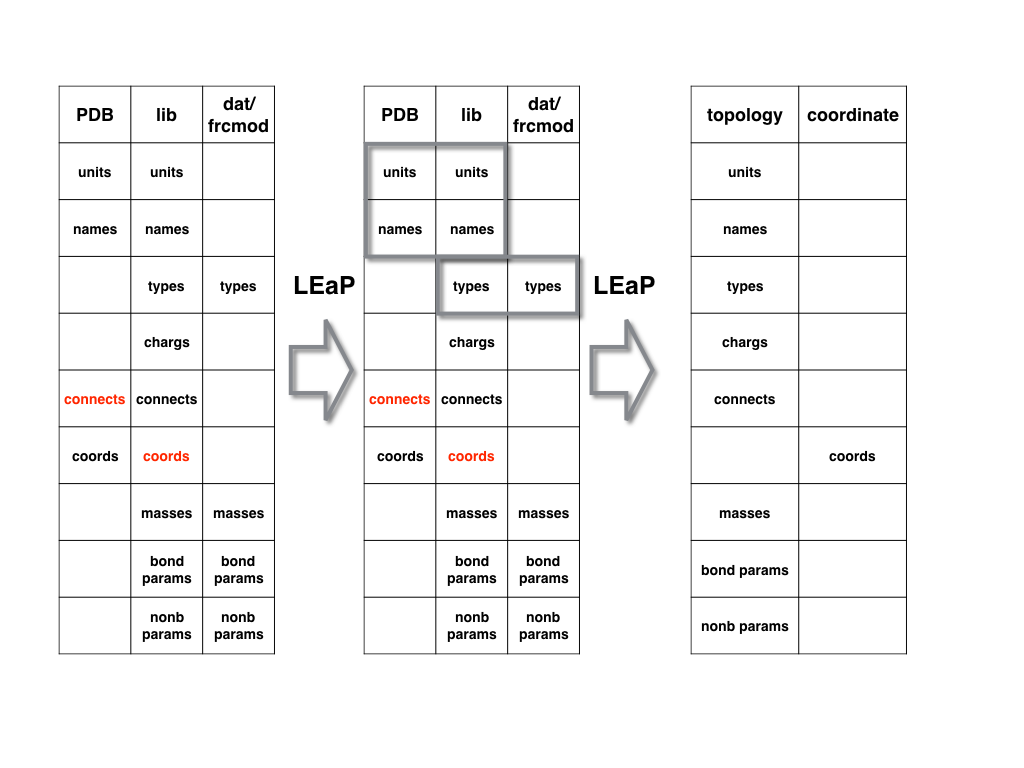
A. For standard residues
For the protein and nucleic acid PDB files, the manner in which LEaP generates
AMBER topology file and coordinate file is shown in the following figure.
AMBER already has lib files for standard amino acids and nucleic acids.
LEaP can merge the information in the PDB and lib files together by first
matching residue names and then matching atom names (it is a one-to-one matching
of each atom in each residue. If there are any missing atoms, LEaP will add it
automatically).
Meanwhile it will merge the information in the lib and dat/frcmod files by
matching the atom type information. And then it will generate the topology and
coordinate files.
B. For non-standard residues
There are no lib files for non-standard residues (such as a ligand, or a rare
amino acid/nucleic acid) in the AMBER force field.
Under this situation users need to created a corresponding "lib file" by
themselves.
Users can create a mol2 or prep (prepi or prepc) file by using antechamber program
or a lib file based on LEaP program.
The former strategy is recommended due to antechamber could deal with that
automatically.
Meanwhile, since the standard AMBER force field may not contain all the force field
parameters needed for the non-standard residue, an additional frcmod file could
be generated using parmchk/parmchk2 program based on the mol2/prep file.
By default, the frcmod file uses General Amber Force Field atom types while
AMBER atom types could also be chosen based on user's choice.
As shown in below, a PDB file could be used to generate prep/mol2 file.
Based on the mol2/prep file, one can use parmchk/parmchk2 to generate the additional
frcmod file.
A prep or mol2 file is recommended to be used as the intermediate file because
they containabundant information and could be used in LEaP program directly.
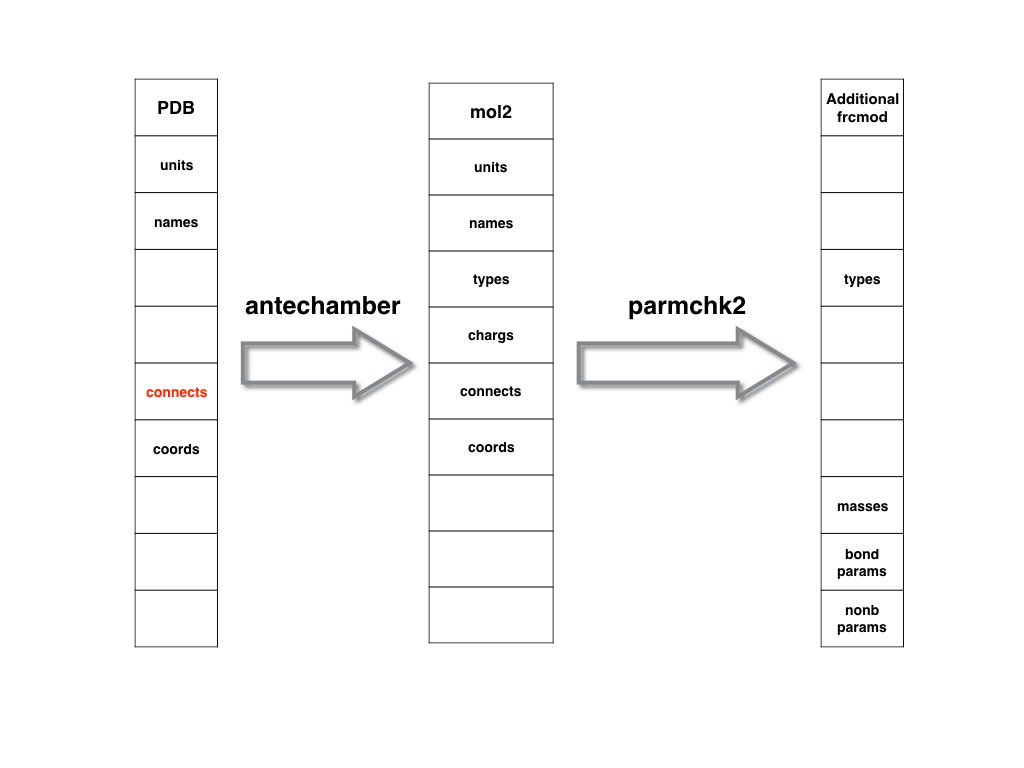
After you obtaining the prep/mol2 and additional frcmod files, you can use them
to generate the top (prmtop) and crd (inpcrd) files by using LEaP.
The flow-chart is shown below.
Error Messages in LEaP
Even advanced AMBER users may meet some problems during the modeling process.
The error messages in the LEaP output and log files could be very helpful at
this time.
Each warning or error usually corresponds to a specific part of atomic
information.
Above figures would be helpful for the user to locate errors and solve problems.
Example
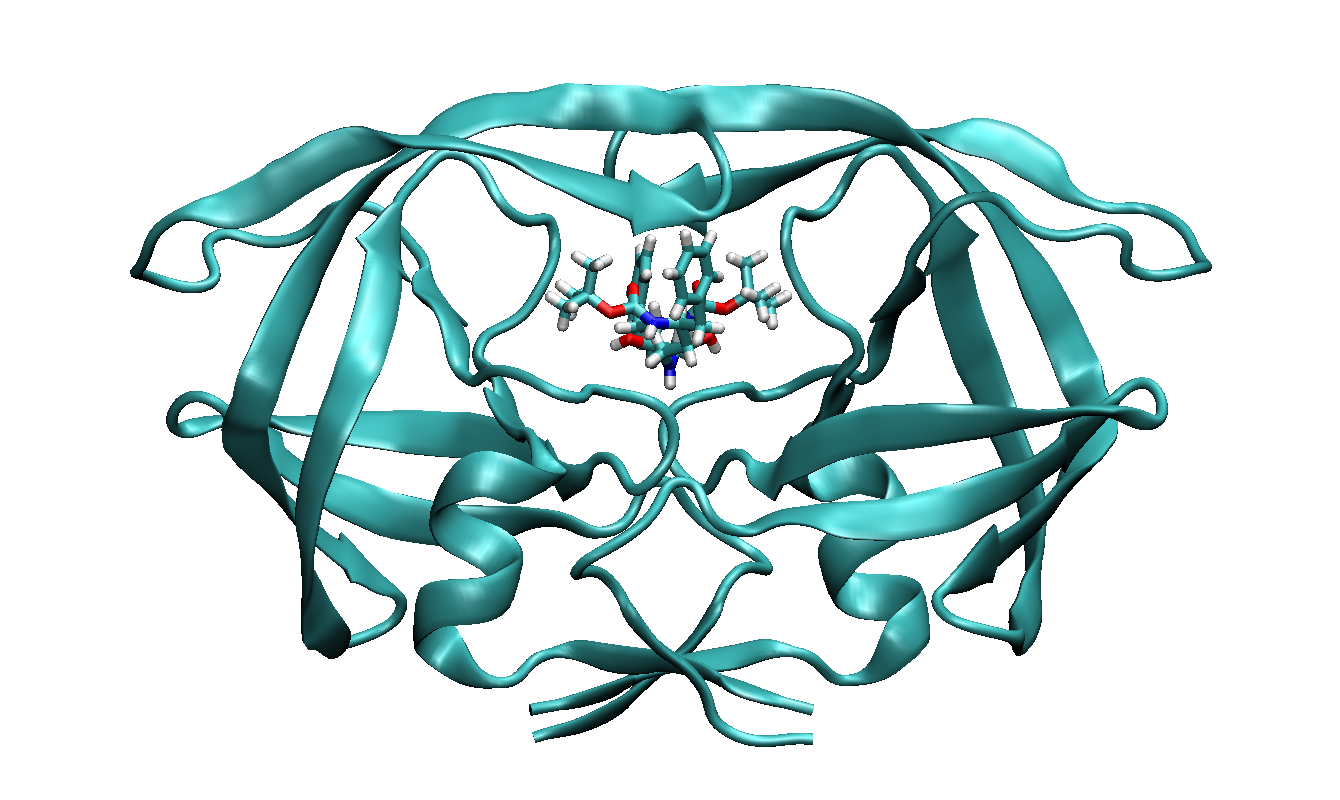
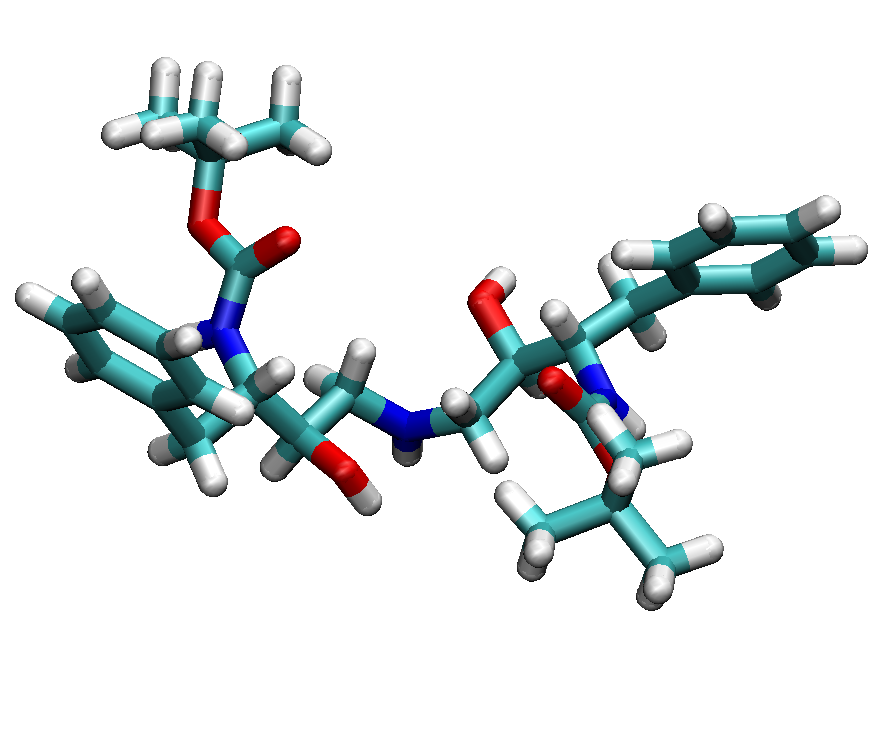
Here we use the structure from PDB entry 1ODX as an example.
It is a ligand bound HIV-Proteinase mutant (A71T, V82A).
The ligand has a chemical formula of
C30H45N3O6 and residue name "0E8".
We have shown the protein-ligand complex and single ligand structures in
following figures.
1. Add hydrogen atoms to the protein system using H++ web-server.
H++ web-server will delete the ligand and only add hydrogen atoms to the protein.
Here are the generated topology and coordinate files of the protein system:
0.15_80_10_pH6.5_1ODX.top,
0.15_80_10_pH6.5_1ODX.crd
2. Generate the protein PDB file using ambpdb program.
We choose this strategy rather than using the PDB file from H++ web-server
directly because all the residues in the topology file have AMBER style
residue names (e.g. all HIS residues have been renamed to HIE, HID or HIP).
Therefore manual changes are not necessary for the PDB file generated
in this way.
ambpdb -p 0.15_80_10_pH6.5_1ODX.top -c 0.15_80_10_pH6.5_1ODX.crd >
0.15_80_10_pH6.5_1ODX.pdb
3. Add hydrogen atoms to the ligand.
We have three commands in this step.
They are for deriving the ligand structure from PDB file with awk, adding
hydrogen atoms to the ligand using reduce (note that there may be wrong
protonation of the structure, under which situation manual fix is necessary),
and cleaning the PDB file by pdb4amber, respectively.
Care should be taken because sometimes hydrogen is added incorrectly by
reduce, for which a manual fix is needed.
awk '$4=="0E8"' 1ODX.pdb > 0E8.pdb
reduce 0E8.pdb > 0E8_H.pdb
pdb4amber -i 0E8_H.pdb -o 0E8_clean_H.pdb
4. Generate a mol2 file and an additional frcmod file for the 0E8 ligand.
First we use antechamber to generate the mol2 file.
Here AM1-BCC charge is used due to its automatic implementation in antechamber
and low computational cost.
Afterwards we use parmchk2 program to construct an additional frcmod file for
the ligand.
antechamber -fi pdb -fo prepi -i 0E8_clean_H.pdb -o 0E8_clean_H.prepi -rn 0E8 -c bcc -pf y
parmchk2 -f prepi -i 0E8_clean_H.prepi -o 0E8.frcmod
5. Combine the protein and ligand PDB files using a cat command and clean the
format by pdb4amber.
cat 0.15_80_10_pH6.5_1ODX.pdb 0E8_clean_H.pdb > 1ODX_H.pdb
pdb4amber -i 1ODX_H.pdb -o 1ODX_clean_H.pdb
6. Use tleap to generate topology and coordinate files for the protein-ligand system.
xleap could also be used for user's preference.
tleap -s -f 1ODX_tleap.in > 1ODX_tleap.out
Here is the content of the 1ODX_tleap.in file:
source leaprc.protein.ff14SB #Source leaprc file for ff14SB protein force field
source leaprc.gaff #Source leaprc file for gaff
source leaprc.water.tip3p #Source leaprc file for TIP3P water model
loadamberprep 0E8_clean_H.prepi #Load the prepi file for the ligand
loadamberparams 0E8.frcmod #Load the additional frcmod file for ligand
mol = loadpdb 1ODX_clean_H.pdb #Load PDB file for protein-ligand complex
solvatebox mol TIP3PBOX 8 #Solvate the complex with a cubic water box
addions mol Cl- 0 #Add Cl- ions to neutralize the system
saveamberparm mol 1ODX.prmtop 1ODX.inpcrd #Save AMBER topology and coordinate files
quit #Quit tleap program
|
The leaprc.protein.ff14SB and leaprc.gaff files are under $AMBERHOME/dat/leap/cmd/
directory.
Comments inside these files can help us know which files have been loaded.
Souring leaprc.protein.ff14SB file will load dat/frcmod files as
parm10.dat, frcmod.ff14SB,
and lib files as amino12.lib (for normal amino acid residues),
aminoct12.lib (for C-terminal amino acid residues),
aminont12.lib (for N-terminal amino acid residues),
Sourcing leaprc.gaff file will load the gaff.dat file.
Sourcing leaprc.water.tip3p will load the lib files as solvents.lib and
atomic_ions.lib, and the frcmod files for atomic ions in TIP3P water.
All the dat/frcmod files are available in $AMBERHOME/dat/leap/parm/ directory,
while all the lib files are under $AMBERHOME/dat/leap/lib/ directory.
Note that the names of leaprc files for different force fields and frcmod files
for ions may change between different AMBER versions, hence
some commands in above input file may need to be changed
or deleted.
Users can check the Specifying which force field you want in LEaP and
Ions sections in the Molecular mechanics force fields chapter
in the corresponding AMBER manual and $AMBERHOME/dat/leap/cmd/ directory for
more details.
Here are the generated topology and coordinate files for the protein-ligand complex:
1ODX.prmtop,
and 1ODX.inpcrd.
Users can use "printBonds", "printAngles", "printDihedrals", and "printDetails" these
commands in ParmEd (see section 14.2 of the
AMBER 2017 Manual)
to check the generated topology and coordinate files.
If all things are good, these files could be used to carry out minimization and then
molecular dynamics simulations.
There is a tutorial about
using the above AMBER force field in CHARMM.
In this way users can take advantage of the functions provided by the CHARMM
software package.
Interested users can check this link.