- 1. Abstract
- 2. Introduction
- 3. Function Form
- 4. Atom Type
- 5. Charge method
- 6. Parameterization for bond length
- 7. Parameterization for bond angle
- 8. Parameterization for torsional angle
- 9. Three test cases
- 10. Summary
1. ABSTRACT
In this work, we have developed a general AMBER force field (GAFF) for rational drug design. GAFF is compatible with the AMBER force field and it has parameters for almost all the organic molecules made of C, N, O, H, S, P, F, Cl, Br and I. As a complete force field, GAFF is suitable for study of a great number of molecules (such as database searching) in an automatic fashion.2. INTRODUCTION
Molecular mechanics are the key component in the armamentarium used by computational chemists for rational drug design and many other tasks. Force fields are the cornerstone of molecular mechanics. A successful force field in drug design should work well both for the biological molecules and the organic molecules. AMBER force fields have a good reputation for the study of proteins and nucleic acids. However, the fact that AMBER only has limited parameters for organic molecules prevents it from being widely used in drug design. Therefore, it is necessary to develop a general AMBER force field that works for most pharmaceutical molecules. It should also be as compatible as possible with the traditional AMBER force fields. Our objective is to develop a general, complete, and compatible force field for rational drug design.3. FUNCTION FORM
Similar to the other AMBER force fields, the GAFF also applies the following simple harmonic functional form: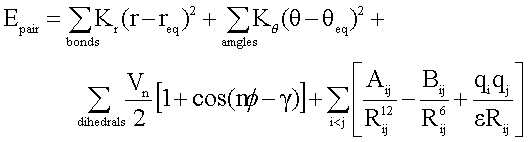
Here, req and qeq are equilibrium structural
parameters; Kr,
Kq,
Vn
are force constants; n is multiplicity and gamma is phase angle for torsional
angle parameters. Details will be given below on how these parameters were
derived.
4. ATOM TYPE DEFINATION
Compared to traditional AMBER force field, atom types in GAFF are more general and cover most of the organic chemical space.Table I lists the basic (a) and special (b) atom types in GAFF.Table I (a). Basic Atom Types in GAFF
| Atom type | Description | Atom type | Description |
| c
c1 c2 c3 ca |
sp2 C in C=O, C=S
sp1 C sp2 C, aliphatic sp3 C sp2 C, aromatic |
o
oh os |
sp2 O in C=O, COO-
sp3 O in hydroxyl group sp3 O in ether and ester |
| n
n1 n2 n3
no |
sp2 N in amide
sp1 N sp2 N with 2 subst. readl double bond sp3 N with 3 subst. sp3 N with 4 subst. sp2 N with 3 subst amine N connected to the aromatic rings N in nitro group |
s2
sh ss s4 s6 |
sp2 S (p=S, C=S etc)
sp3 S in thiol group sp3 S in -SR and SS hypervalent S, 3 subst. hypervalent S, 4 subst. |
| hc
ha hn ho hs hp |
H on aliphatic C
H on aromatic C H on N H on O H on S H on P |
||
| f
cl br i |
any F
any Cl any Br any I |
p2
p3 p4 p5 |
sp2 P (C=P etc)
sp3 P, 3 subst. hypervalent P, 3 subst. hypervalent P, 4 subst. |
Table I (b). Special Atom Types in GAFF
| Atom type | Description | Atom type | Description |
| h1
h2 h3 h4 h5 |
H on aliphatic C with 1 EW group;
H on aliphatic C with 2 EW group; H on aliphatic C with 3 EW group; H on aromatic C with 4 EW group; H on aromatic C with 5 EW group; |
cc(cd)
ce(cf) cp(cq) cu cv cx cy |
inner sp2 C in conj. ring systems
inner sp2 C in conj. chain systems bridge aromatic C sp2 C in three-memberred rings sp2 C in four-memberred rings sp3 C in three-memberred rings sp3 C in four-memberred rings |
| n
nb nc(nd) sx sy |
aromatic nitrogen
inner sp2 N in conj. ring systems inner sp2 N in conj. chain systems conj. S, 3 subst. conj. S, 4 subst. |
pb
pc(pd) pe(pf) px py |
aromatic phosphorus
inner sp2 P in conj. ring systems inner sp2 P in conj. chain systems conj. P, 3 subst. conj. P, 4 subst. |
5. CHARGE APPROACH
The charge method used in GAFF is HF/6-31G* RESP charge. However, AM1-BCC, which was parameterized to reproduce the HF/6-31G* RESP charges can be applied in case of large amount of calculations, such as database searching. The van der Waals parameters of GAFF are as same as those used by the traditional AMBER force field.6. BOND LENGTH PARAMETERIZATION
There are several resources to derive the reference bond length req, including the statistic mean values of bond lengths from X-ray and neutron diffraction as well as high level ab initio calculations (MP2/6-31G* ). Force constants were derived using the following empirical function forms: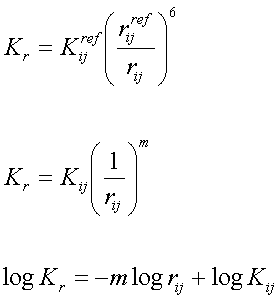
Here, m and Kij were determined using bond length parameters in traditional AMBER force field. As a compromise, the parameter m in the above equation is set to 4.0. Fitting details are shown in Figure 1-3. The parameters are listed in Table II.
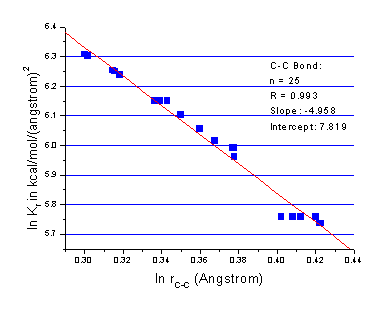
Figure 1
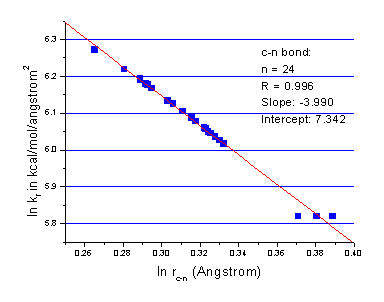
Figure 2
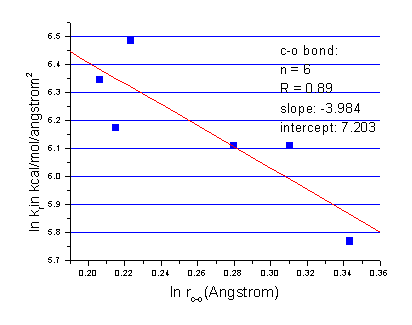
Figure 3
Table II. Bond length parameterization
| i | j | rij | Kij | i | j | rij | Kij |
| H | H | 0.738 | 4.661 | H | C | 1.090 | 6.217 |
| H | N | 1.010 | 6.057 | H | O | 0.96 | 5.794 |
| H | F | 0.920 | 5.600 | H | Cl | 1.280 | 6.937 |
| H | Br | 1.410 | 7.301 | H | I | 1.600 | 7.802 |
| H | P | 1.410 | 7.257 | H | S | 1.340 | 7.018 |
| C | C | 1.526 | 7.643 | C | N | 1.470 | 7.504 |
| C | O | 1.440 | 7.347 | C | F | 1.370 | 7.227 |
| C | Cl | 1.800 | 8.241 | C | Br | 1.940 | 8.478 |
| C | I | 2.160 | 8.859 | C | P | 1.830 | 8.237 |
| C | S | 1.820 | 8.117 | N | N | 1.441 | 7.634 |
| N | O | 1.420 | 7.526 | N | F | 1.420 | 7.475 |
| N | Cl | 1.750 | 8.266 | N | Br | 1.930 | 8.593 |
| N | I | 2.120 | 8.963 | N | P | 1.720 | 8.212 |
| N | S | 1.690 | 8.073 | O | O | 1.460 | 7.561 |
| O | F | 1.410 | 7.375 | O | Cl | 1.700 | 8.097 |
| O | Br | 1.790 | 8.276 | O | I | 2.110 | 8.854 |
| O | P | 1.640 | 7.957 | O | S | 1.650 | 7.922 |
| F | F | 1.406 | 7.358 | F | Cl | 1.648 | 7.947 |
| F | P | 1.500 | 7.592 | F | S | 1.580 | 7.733 |
| Cl | Cl | 2.031 | 8.648 | Cl | I | 2.550 | 9.309 |
| Cl | P | 2.040 | 8.656 | Cl | S | 2.030 | 8.619 |
| Br | Br | 2.337 | 9.012 | Br | I | 2.671 | 9.380 |
| Br | P | 2.240 | 8.729 | Br | S | 2.210 | 8.728 |
| I | I | 2.836 | 9.511 | I | P | 2.490 | 9.058 |
| I | S | 2.560 | 9.161 | P | P | 2.324 | 8.805 |
| P | S | 2.120 | 8.465 | S | S | 2.038 | 8.316 |
7. BOND ANGFGLE PARAMETERIZATION
The following list the source of reference bond angles:
- Cambridge Structure Database (CSD)
- ab initio (MP2/6-31G*)
- empirical rules for q (A-B-C)
q(A-B-C) = 0.5 [q (A-B-A)+ q (C-B-C)]
The force constant Kqwas also estimated using
an empirical function as the following. The parameters are listed in Table
III.
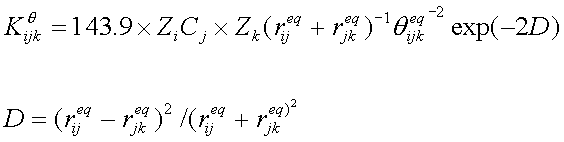
Table III. Parameters of bond angle force constant calculations
| Element | C | Z |
| H | 0.0 | 0.784 |
| C | 1.339 | 1.183 |
| N | 1.300 | 1.212 |
| O | 1.249 | 1.219 |
| F | 0.000 | 1.166 |
| Cl | 0.000 | 1.272 |
| Br | 0.000 | 1.378 |
| I | 0.000 | 1.398 |
| P | 0.906 | 1.620 |
| S | 1.448 | 1.280 |
8. TORSIONAL ANGLE PARAMETERIZATION
The following is the strategy to develop torsional angle parameters.- Perform torsional angle scanning to get the rotational profile at MP4/6-311G(d,p)// MP2/6-31G* level
- Apply PARMSCAN to find the best torsional angle parameters to reproduce the rotational profile
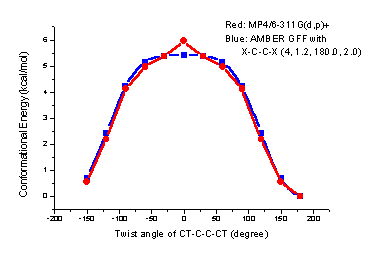
Figure 4. An example: rotation profiles of X-c-c-X
9. TEST CASES
Table IV Comparison of GAFF and other force fields in reproducing geometries of
75 crystallographic structures
| GAFF | MMFF94 | DREIDING | TRIPOS 5.2 | CHARMm | |
| RMS of Atomic Displacement (Å) | 0.34 | 0.47 | 0.24 | 0.25 | 0.44 |
| RMS of Bond Length Deviation (Å) | 0.027 | 0.021 | 0.035 | 0.025 | ~ |
| RMS of Bond Angle Deviation (°) | 2.2 | 2.0 | 3.22 | 2.5 | ~ |
Table V How well does GAFF perform in calculating the inter-molecular
energies
of 22 base pairs. All the comparison are made to the MP2/6-311G* energies.
| GAFF/BCC | GAFF/RESP | PARM99/RESP | |
| RMS of Atomic Displacements (Å) | 0.56 | 0.49 | 0.19 |
| RMS of DE Compared to ab initio (kcal/mol) | 2.50 | 0.67 | 0.77 |
Table VI Performances of widely used force field in reproducing
the relative
energies for 55 compounds. Comparison are made to the experiment.
| GAFF/BCC | AMBER (parm99) | MMFF | MM3 | CHARMm | |
| Unsigned Average Error (kcal/mol) | 0.51 | 0.28 | 0.43 | 0.52 | 0.57 |
| RMS Deviation (kcal/mol) | 0.69 | 0.40 | 0.54 | 0.75 | 0.76 |
10. SUMMARY
In this work, we have developed a general AMBER force field. We hope it is an ideal molecular mechanical tool in rational drug design. We have finished a total of 2000 MP2/6-31G* optimizations and 1260 MP4/6-311G(d,p) single point calculations. From the three test cases, encouraging results were achieved. It is notable that in the third test case, GAFF has comparable performance to those of CHARMm and MM3, although GAFF applies very general force field parameters and crude charge approach.Compared to other widely used fields, GAFF has the following distinguishing features. First of all, it is a complete force field, which means all the parameters are available, no parameter-missing happens to GAFF; secondly, this force field is very general and it covers almost all the organic chemical spaces; finally, GAFF is compatible to the AMBER force field. We believe that the combination of GAFF with traditional AMBER will offer a useful molecular mechanical tool for rational drug design, especially for things like binding free energy calculations.