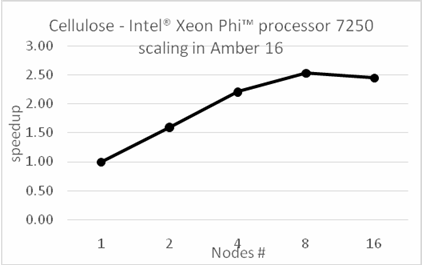
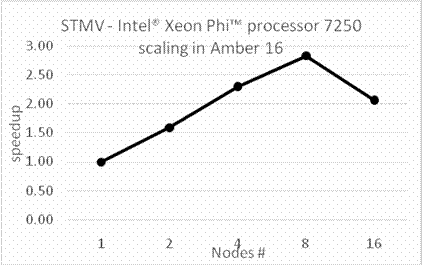
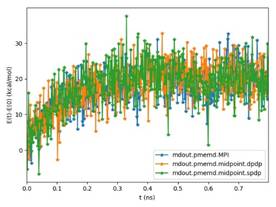
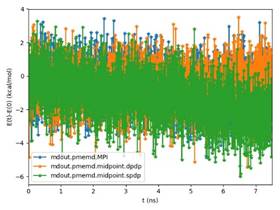
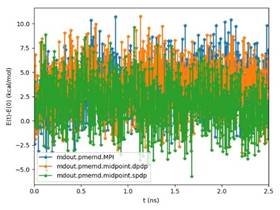
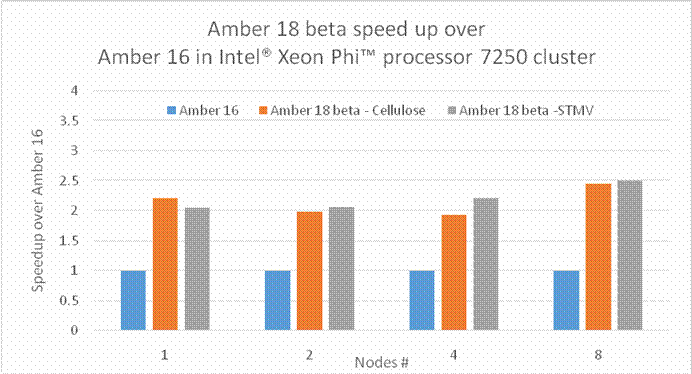
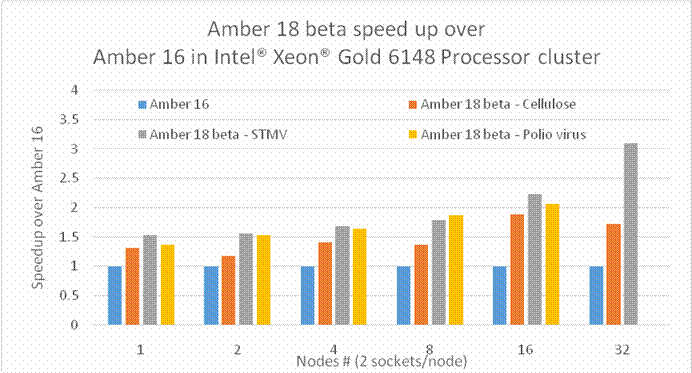
BackgroundAbout the new Midpoint method implementation in Amber 18The Midpoint method for Molecular Dynamics was developed by researchers at IBM as part of their BlueMatter project with BlueGene and subsequently expanded upon and described publically by researchers at D. E. Shaw Research, LLC (see this 2006 paper published in The Journal of Chemical Physics). The Midpoint method is based on domain decomposition and provides anefficient approach to significantly reduce “data distribution” time with increased node count. The San Diego Computing Center (SDSC) at UC San Diego in collaboration with Intel© Corporation have developed a prototype implementation of the Midpoint method within the Amber PMEMD software. This implementation has been thoroughly tested with three key Amber benchmarks. The midpoint implementation will continue to be refined and the performance improved over the coming months through patches in Amber 18. Motivation for re-architecting Amber 16 PMEMD CPU codeThe aim to re-architect Amber 16’s PMEMD code is to continue to improvethe CPU performance and cluster scalability. For example, the following figures show thatthe current atom decomposition approach in Amber 16 does not scale well for theCellulose andSTMV benchmarks (roughly 400K and 1 Million atoms, respectively) beyond 8 nodes each containing a single Intel© Xeon Phi™processor 7250 (Knights Landing). It was also observed that the data distribution time grew exponentially with increased node count.
Figure 1: Scaling of existing Amber 16 PMEMD code on Intel Xeon Phi 7250 equipped nodes connected with Intel Omni Path.
Authorship & SupportPMEMD Midpoint Implementations:
Citing the Midpoint Code If you make use of any of the Amber midpoint code in your work please include the following citations (in addition to the standard Amber citation):
|
|