The two ligands were sketched and parametrized with gaff atom types and resp charges were generated using antechamber on gaussian03 output files. (Please refer to tutorial 4 on how to use the antechamber tools to parametrize a ligand. The benzene and phenol molecules were saved in two OFF-libraries (benz.lib and phen.lib) for further use. The screenshot shows that C6 was selected as the position bearing the hydroxyl group in phenol.
We will use the X-ray structure of T4-L99A from the pdb (after stripping water
molecules and unneeded heteroatoms from it: pdb
file) as basis to set up our simulation files. We will use two runs of leap
to produce four set of parameter and restart files, containing both ligands in
the protein bound and solvated states. The first leap run (input file) will produce pdb files of the solvated und
neutralized benzene complex and of the benzene ligand in water (complex.pdb and ligand.pdb). From these two additional pdb files are made
by renaming the BNZ molecule to PHN and deleting H6 (t4_phn.pdb and phn.pdb). These four
pdb files are then used in a second leap run (input
file) to generate the *.prm and *.rst files. This yields 4 parameter and 4
rst files
| benzene | phenol | |
| complex | prm/rst | prm/rst |
| solvated ligand | prm/rst | prm/rst |
Two different ligand transformations need to be simulated, benzene to phenol in water and bound to lysozyme (Simulating a 'back' transformation phenol to benzene is not necessary, TI calculations do not have a direction, rather every λ point represents a static intermediate of the transformation). For reasons of simulation consistency, the two transformations are further broken up into three substeps each: First, the atomic partial charge on the hydrogen atom to disappear from benzene are removed, second the ligand vdW-transformation is performed (disappearing or decoupling the benzene hydrogen from its surroundings while simultaneously appearing the phenol hydroxyl group) and finally the phenol hydrogen group gets its atomic partial charges switched on. This is done because having a nonzero charge on an atom while the vdW interactions with its surroundings are getting weakers can lead to simulation instabilities. Also for reasons of simulation consistency, in the van-der-Waals transformation, we will use softcore potentials, a modified LJ-equation that prevents the origin singularity type of free energy divergence from happening (see Simonson T, Mol. Phys 1993: 80:441pp for more information).
The mdin files used for these transformations contain the normal Amber parameters for performing Minimizations/MD simulations that I will not discuss further and in addition to that some unique parameters to perform the TI calculations. The non-TI parts of the input files will perform the following steps:
| Input file | Step 1 (9 λ points:
0.1,0.2...0.9) (removing charge on BNZ H6) | Step 2 (9 λ points:
0.1,0.2...0.9) (changing BNZ into PHN) | Step 3 (9 λ points:
0.1,0.2...0.9) (Adding charges on PHN O1,H6) |
| Process 0 (V0) |
|
|
|
| Process 1 (V1) |
|
|
|
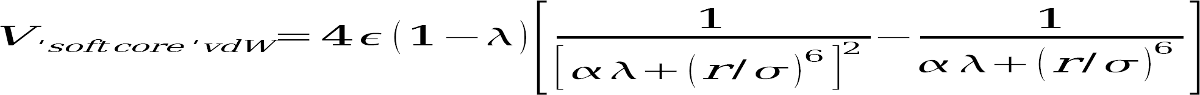
Setting ifsc=1
also informs sander that a number of atoms in each prmtop file (those specified
by scmask) shall be considered unique to this potential function. In the step 1
and 3 transformations, we do not need to use softcore potentials, as the only
difference between V0 and V1 is the presence or absence of some partial
charges, while both start- and endstate have the same number of atoms.
Actually running the simulations will use the multisander functionality of
sander.MPI, using e.g.: 'mpirun -np 2 sander.MPI -ng 2 -groupfile group'. Due to the multitude of different input files, it is
very advisable to use some sort of script to automate their generation. For
example, a simple shell script like this,
which sets up the charge removal TI run for benzene in the complex, could serve
as a starting point. The script generates a pbs input file that you can adjust
for your own brand of supercomputer queueing system. So in the end, a total of
two transformations, with three substeps each, with nine λ windows each
with three simulations each will be performed, for a total of 162 invocations
of the sander module (each using at least two CPUs). A real TI study might
involve many more λ windows and significantly longer simulation times,
making this a very computationally intensive technique.
This tutorial will continue with a description of running the simulations and analyzing the results here
{kind=link}