This page is an archive of AMBER 14 GPU support.
For the most up to date GPU support in AMBER refer to the primary GPU
page here.
News and Updates
Dec 2015: Exxact
AMBER Certified GPU Solutions are now shipping with an optimized
version (developed by Ross Walker in conjunction with Exxact and
NVIDIA) of the AMBER v14 software that provides ~15% faster
performance on comparable, NVIDIA Maxwell [GTX980,
980TI,
Titan-X, M40 and M60], hardware with bitwise identical results.
All Exxact AMBER
Certified GPU systems shipped after Dec 20th 2015 include this
custom optimized version of AMBER v14 preinstalled as well as free
support for upgrading to AMBER v16 once it is released [AMBER 16
license required]. See benchmark
page for performance comparison.
Jul 2015: Validated and warranted GTX780Ti
systems available from Exxact at heavily discounted price. (SOLD
OUT)
[Custom bios solves 780Ti hardware issues allowing them to be used
with AMBER].
Jul 2015:
GTX980Ti GPU now validated for numerical accuracy and
benchmarked.
Jan 2015: Amber Update.10 adds support for Center
of Mass restraints to the GPU code.
Nov 2014: Added benchmark numbers for NVIDIA's
latest Tesla part: K80.
Oct 2014: Support for NVIDIA
Maxwell cards (aka GTX980/970) released as part of update.16 to
AmberTools. This requires a minimum of CUDA 6.5.
Sept 2014:New recommended hardware designs announced. High Performance - 8
GPU systems and High Density (2U) 4 GPU systems added to the list of
Exxact AMBER Certified GPU Computing solutions.
Apr 2014: AMBER 14 released. >30% performance
improvement, peer to peer parallel support and many new GPU features
added.
Aug 2013: In collaboration with
Exxact Corp we have put together an updated version of the
AMBER Certified MD Workstation program. This provides a simple
way to purchase machines for running GPU accelerated AMBER. The
systems come with AMBER 12 or 14 pre-installed (AMBER license required)
and fully tested and are ready to run out of the box. A range of
AMBER optimized specifications are available for different budget
levels and all are fully customizable and include a full warranty.
Free test drives of these machines are also available. (More details
are available on the Recommended
Hardware Page.)
One of the new features of AMBER, introduced with version 11, was
the ability to use NVIDIA GPUs to massively accelerate PMEMD for both explicit
solvent PME and implicit solvent GB simulations. This was further extended in AMBER 12
and the performance envelope has been pushed even further
with AMBER 14. This work is by Ross Walker at the San Diego
Supercomputer Center and Scott Le Grand at Amazon Web
Services in collaboration with NVIDIA. While this GPU
acceleration is considered to be production ready, and has
been heavily used since it's release as part of AMBER 11, it
is still evolving and thus has not been tested to the same
extent as the CPU code has over the years and so users
should still exercise caution when using this code. The
error checking is not as verbose in the GPU code as it is on
the CPU. In particular simulation failures, such as atom
collisions or other simulation instabilities will manifest
themselves as CUDA launch errors or GPU download failures
and not as informative error messages. If you encounter
problems during a simulation on the GPU you should first try
to run the identical simulation on the CPU to ensure that it
is not your simulation setup which is causing problems. That
said the AMBER GPU code is in use in hundreds of research
labs worldwide. If you want a simple turnkey ready to run
GPU workstation or cluster with AMBER preinstalled (and
optimized for a ~15% performance boost) please
see the AMBER
Certified GPU solutions on the recommended hardware page.
If you
encounter problems during a simulation on the GPU you should
first try to run the identical simulation on the CPU to
ensure that it is not your simulation setup which is causing
problems. Feedback and questions should be posted to the
Amber
mailing list.
New in AMBER 14
AMBER 14 includes full GPU support in
PMEMD and is a major update over AMBER 12. Focus for AMBER
14 has been on extending the performance envelope,
supporting multi-GPU runs efficiently and further increasing the features available in the GPU
accelerated code. Key new features in the AMBER 14 GPU version of PMEMD include:
~30% performance improvement for single GPU runs.
Addition of peer to peer support for multi-GPU runs
providing enhanced multi-GPU scaling.
Hybrid bitwise reproducible fixed point precision
model as standard (SPFP)
Support for Extra Points in Multi-GPU runs.
Jarzynski Sampling
GBSA support
Support for off-diagonal modifications to VDW
parameters.
Multi-dimensional Replica Exchange (Temperature and
Hamiltonian)
Support for CUDA 5.0, 5.5, 6.0, 6.5 and 7.0
Support for latest generation GPUs.
Monte Carlo barostat support providing NPT
performance equivalent to NVT.
ScaledMD support.
Improved accelerated (aMD) MD support.
Explicit solvent constant pH support.
NMR restraint support on multiple GPUs.
Improved error messages and checking.
Hydrogen mass repartitioning support (4fs time
steps).
Support for latest generation Maxwell GPUs (GTX9XX,
GTX-Titan-X)
Support is provided for single GPU and multiple GPU runs.
Employing multiple GPUs in a single simulation requires MPI
and the pmemd.cuda.MPI executable. If you have multiple
simulations to run then the recommended method is to use one
GPU per job. The pmemd GPU code has been developed in such a
way that for single GPU runs the PCI-E bus is only used for
I/O. This sets AMBER apart from other MD packages since it
means the CPU specs do not feature in the GPU code
performance. As such low end economic CPUs can be used.
Additionally it means that in a system containing 4 GPUs 4
individual calculations can be run at the same time without
interfering with each other's performance.
Further information relating to the
specifics of the implementation, methods used to achieve
performance while controlling accuracy, and details of
validation are available or will be shortly from the following publications:
Romelia Salomon-Ferrer;
Andreas W. Goetz; Duncan Poole; Scott Le Grand; &
Ross C. Walker* "Routine microsecond molecular
dynamics simulations with AMBER - Part II: Particle Mesh
Ewald" , J. Chem. Theory Comput., 2013,
9 (9), pp 3878-3888 DOI:
10.1021/ct400314y
Andreas W. Goetz; Mark
J. Williamson; Dong Xu; Duncan Poole; Scott Le Grand; & Ross C. Walker* "Routine
microsecond molecular dynamics simulations with AMBER -
Part I: Generalized Born", J. Chem. Theory
Comput., 2012, 8 (5), pp 1542-1555, DOI:
10.1021/ct200909j
Scott Le Grand; Andreas
W. Goetz; & Ross C. Walker* "SPFP: Speed
without compromise - a mixed precision model for GPU
accelerated molecular dynamics simulations.",
Comp. Phys. Comm, 2013, 184, pp374-380, DOI:
10.1016/j.cpc.2012.09.022
Funding for this work has been graciously
provided by
NVIDIA, The University of California (UC Lab
09-LR-06-117792), The
National
Science Foundation's (NSF) TeraGrid Advanced User Support
Program through the
San Diego
Supercomputer Center and NSF SI2-SSE grants to Ross
Walker (NSF1047875 / NSF1148276) and Adrian Roitberg (NSF1047919
/ NSF1147910)
Citing the GPU Code
If you make use of any of this GPU support in
your work please use the following citations (in addition to
the standard AMBER 14 citation):
Explicit Solvent
Romelia Salomon-Ferrer;
Andreas W. Goetz; Duncan Poole; Scott Le Grand; &
Ross C. Walker* "Routine microsecond molecular
dynamics simulations with AMBER - Part II: Particle Mesh
Ewald" , J. Chem. Theory Comput., 2013,
9 (9), pp 3878-3888. DOI:
10.1021/ct400314y
Implicit Solvent
Andreas W. Goetz; Mark
J. Williamson; Dong Xu; Duncan Poole; Scott Le Grand; & Ross C. Walker* "Routine
microsecond molecular dynamics simulations with AMBER -
Part I: Generalized Born", J. Chem. Theory
Comput., 2012, 8 (5), pp 1542-1555 , DOI:
10.1021/ct200909j
The GPU accelerated version of PMEMD
14, supports both explicit solvent PME or IPS simulations in all three canonical ensembles (NVE, NVT
and NPT) and implicit solvent Generalized Born simulations.
It has been designed to support as many of the standard
PMEMD v14 features as possible, however, there are some
current limitations that are detailed below. Some of these
may be addressed in the near future, and patches released,
with the most up to date list posted on the web page. The
following options are NOT supported (as of the
AMBER GPU v14.0.1 release):
1) ibelly /= 0
Simulations using
belly style constraints are not supported.
2) icfe /= 0
Support for
TI is not currently implemented. It is hoped
that an update will be released in a few months
that adds support for TI.
3) if (igb/=0 &
cut<systemsize)
GPU accelerated
implicit solvent GB simulations do not support a
cutoff.
4) nmropt > 1
Support is not
currently available for nmropt > 1. In addition
for nmropt = 1 only features that do not change
the underlying force field parameters are
supported. For example umbrella sampling
restraints are supported as well as simulated annealing
functions such as variation of Temp0 with
simulation step. However, varying the VDW
parameters with step is NOT supported.
5) nrespa /= 1
No multiple time stepping is supported.
6) vlimit /= -1
For performance
reasons the vlimit function is not implemented
on GPUs.
7) es_cutoff /=
vdw_cutoff
Independent cutoffs
for electrostatics and van der Waals are not
supported on GPUs.
8) order > 4
PME interpolation orders of greater than 4 are not supported at present.
9) imin=1 (in
parallel)
Minimization is only
supported in the serial GPU code.
10) netfrc is
ignored
The GPU code does not
currently support the netfrc correction in PME
calculations and the value of netfrc in the
ewald namelist is ignored.
11) emil_do_calc /=
0
Emil is not supported on GPUs.
12) lj264 /= 0
The 12-6-4 potential is not supported on
GPUs.
13) isgld > 0
Self guided langevin is not supported on
GPUs.
13) iemap > 0
EMAP restraints are not supported on
GPUs.
Additionally there are some minor
differences in the output format. For example the Ewald
error estimate is NOT calculated when running on a GPU. It
is recommended that you first run a short simulation using
the CPU code to check the Ewald error estimate is reasonable
and that your system is stable. With the exception of item
10 the above limitations are
tested for in the code, however, it is possible that there
are additional simulation features that have not been
implemented or tested on GPUs.
GPU accelerated PMEMD has been implemented using
CUDA and thus will only run on NVIDIA GPUs at present. Due to accuracy
concerns with pure single precision the code uses a custom designed
hybrid single / double / fixed precision model termed
SPFP. This places
the requirement that the GPU hardware supports both double precision and
integer atomics meaning only GPUs with hardware revision 2.0 and later
can be used. Support for hardware revision 1.3 was present in previous
versions of the code but for code complexity and maintenance reasons has
been deprecated in AMBER 14. At the time of Amber's release the
following cards are supported (* = untested):
Caution: With the exception of Exxact AMBER certified cards the
GTX-780Ti cards are NOT recommended at this time due to instability and
numerical accuracy during AMBER MD simulations which we have tracked
down to a specific hardware design flaw. In collaboration with Exxact we have
developed a custom GTX780Ti bios that circumvents this hardware flaw and
makes the cards fully stable for CUDA workloads.
GTX-Titan and GTX-780 cards require NVIDIA Driver version >= 319.60.
GTX-Titan-Black Edition cards require
NVIDIA Driver version >= 337.09 .or. 331.79 or later.
GTX-970 / 980 / Titan-X cards are supported but require at least CUDA v6.5.
Other cards not listed here may also be supported as long as they
correctly implement the Hardware Revision 2.0, 3.0, 3.5, 5.0 or 5.5
specifications.
Note that you should ensure that all GPUs on which you plan to run
PMEMD are connected to PCI-E 2.0 x16 lane slots or better, especially
when running in parallel across multiple GPUs which really needs PCI-E
3.0 x16 with peer to peer support for reasonable scaling. If this is
not the case then you will likely see degraded
performance, although this effect is lessened in serial if you write to
the mdout or mdcrd files infrequently (e.g. every 2000 steps or so). Scaling over multiple GPUs within a single node is possible, if all
are in x16 or better slots and can communicate via peer to peer [i.e.
connected to the same physical processor socket]. It is also possible to run over multiple
nodes using infiniband but not recommended except for loosely coupled
runs such as REMD. The main advantage of AMBER's approach to GPU
implementation over other implementations such as NAMD and Gromacs is
that it is possible to run multiple single GPU runs on a single node
with little or no slow down. For example a node with 4 GTX-Titan-X cards could run 4
individual AMBER DHFR 4fs NVE calculations all at the same time without
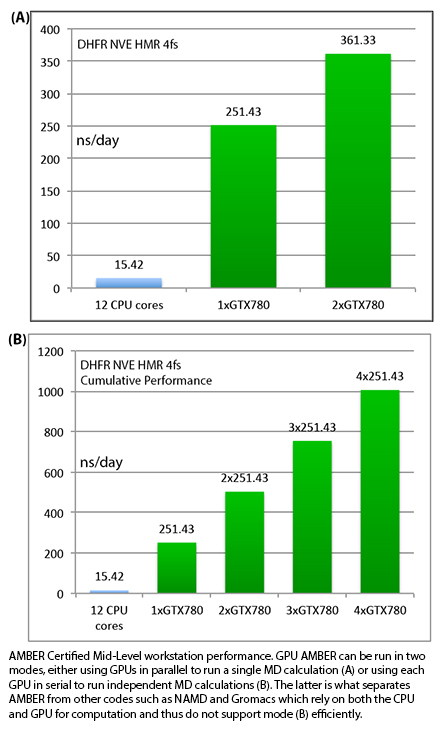
slowdown providing an aggregate throughput in excess of 1250ns/day.
In order to obtain the extensive speedups that we
see with GPUs it is critical that all of the calculation take place on
the GPU within the GPU memory. This avoids the performance hit that one
takes copying to and from the GPU and also allows us to achieve
extensive speedups for realistic size systems. This avoids the need to
create systems with millions of atoms to show reasonable speedups even
when sampling lengths are unrealistic. This unfortunately means that the
entire calculation must fit within the GPU memory. Additionally we make
use of a number of scratch arrays to achieve high performance. This
means that the GPU memory usage can actually be higher than a typical
CPU run. It also means, due to the way we had to initially implement
parallel GPU support that the memory usage per GPU does NOT
decrease as you increase the number of GPUs. This is something we hope
to fix in the future but for the moment the atom count limitations
imposed on systems by the GPU memory is roughly constant whether you run
in serial or in parallel.
Since, unlike CPUs it is not possible to add more
memory to a GPU (without replacing it entirely) and there is no concept
of swap as there is on the CPU the size of the GPU memory imposes hard
limits on the number of atoms supported in a simulation. Early on within
the mdout file you will find information on the GPU being used and an
estimate of the amount of GPU and CPU memory required:
|------------------- GPU DEVICE
INFO --------------------
|
| CUDA Capable Devices Detected: 1
| CUDA Device ID in use: 0
| CUDA Device Name: Tesla C2070
| CUDA Device Global Mem Size: 6143 MB
| CUDA Device Num Multiprocessors: 14
| CUDA Device Core Freq: 1.15 GHz
|
|--------------------------------------------------------
...
| GPU memory information:
| KB of GPU memory in use: 4638979
| KB of CPU memory in use: 790531
The reported GPU memory usage is
likely an underestimate and meant for guidance only to give you an idea
of how close you are to the GPU's memory limit. Just because it is less
than the available Device Global Mem Size does not necessarily mean that
it will run. You should also be aware that the GPU's available memory is
reduced by 1/9th if you have ECC turned on.
Memory usage is affected by the run
parameters. In particular the size of the cutoff, larger cutoffs needing
more memory, and the ensemble being used. Additionally the physical GPU
hardware affects memory usage since the optimizations used are
non-identical for different GPU types. Typically, for PME runs, memory
usage runs:
Use of restraints etc will also
increase the amount of memory in use. As will the density of your
system. The higher the density the more pairs per atom there are and
thus the more GPU memory will be required. The following table provides
an approximate UPPER BOUND to the number of atoms supported as a function of GPU model. These numbers were estimated
using boxes of TIP3P water (PME) and solvent caps of TIP3P water (GB).
These had lower than optimum densities and so you may find you are
actually limited for dense solvated proteins to around 20% less than the
numbers here. Nevertheless these should provide reasonable estimates to
work from.
All numbers are for SPFP
precision and are approximate limits. The actual limits will depend on
system density, simulation settings etc. These numbers are thus
designed to serve as guidelines only.
The nature of current generation GPUs
is such that single precision arithmetic is considerably
faster (>23x for K10, >2x for K20) than double
precision arithmetic. This poses an issue when trying to
obtain good performance from GPUs. Traditionally the CPU
code in Amber has always used double precision throughout
the calculation. While this full double precision approach
has been implemented in the GPU code it gives very poor
performance. Thus the AMBER GPU implementation was initially
written to use a combination of single and double
precision, termed hybrid precision (SPDP), that is discussed
in further detail in the references provided at the
beginning of this page. This
approach used single precision for individual calculations
within the simulation but double precision for all
accumulations. It also used double precision for shake
calculations and for other parts of the code where loss of
precision was deemed to be unacceptable. Tests have shown
that energy conservation is equivalent to the full double
precision code and specific ensemble properties, such as
order parameters, match the full double precision CPU code. The user should understand though that this
approach leads to divergence between GPU and CPU
simulations, similar to that observed when running the CPU
code across different processor counts in parallel but
occurring more rapidly. For this reason the GPU test
cases are more sensitive to rounding differences caused by
hardware and compiler variations and will likely require
manual inspection of the test case diff files in order to
verify that the installation is providing correct results.
There can also be differences between CPU and GPU runs and between GPU and GPU on different GPU models for
runs that rely on the random number stream. For example
NTT=2 and NTT=3. This is because the random number stream is
different between CPUs and GPUs.
With AMBER 14 the single and double
precision models (SPSP, SPDP and DPDP) have been deprecated
and replaced with a new
hybrid model that combines single precision calculation with
fixed precision accumulation. This is described in detail in
the following manuscript and tests have shown that it
provides accuracy that is as good or better than the
original SPDP model. This new precision model is supported
on v2.0 hardware and newer and is especially beneficial on Kepler
I (3.0) hardware.
Scott Le Grand; Andreas
W. Goetz; & Ross C. Walker* "SPFP: Speed
without compromise - a mixed precision model for GPU
accelerated molecular dynamics simulations.",
Comp. Phys. Comm, 2013, 184, pp374-380, DOI: 10.1016/j.cpc.2012.09.022
While the default precision model is
currently the new SPFP model there is also a DPFP model
available which provides full double precision calculation
with bitwise reproducible fixed precision accumulation to
facilitate advanced testing and comparison. The precision models supported
in AMBER 14, and
determined at compile time as described later, are:
SPFP - (Default)
Uses a combination of single precision for calculation
and fixed (integer) precision for accumulation. This
approach provides optimum speedup on Kepler cards,
minimizes memory overhead and provides greater net
precision than the original SPDP model. It is designed for
optimum performance on hardware revision 2.0 or later.
DPFP - Uses double
precision (and double precision equivalent fixed
precision) for the entire calculation. This provides for
careful regression testing against the CPU code. It
makes no additional approximations above and beyond the
CPU implementation and would be the model of choice if
performance was not a consideration. On v2.0 NVIDIA
hardware (e.g. M2090) the performance is approximately
half that of the SPFP model while on v3.0 NVIDIA
hardware (e.g. K10) the performance is substantially
less than the SPFP model.
Recommendation for Minimization
One limitation of the SPFP precision model is that force
can be truncated if they overflow the fixed precision
representation. This should never be a problem during MD
simulations for any well behaved system. However, for
minimization or very early in the heating phase it can
present a problem. This is especially true if two atoms are
close to each other and thus have large VDW repulsions. It
is recommended therefore that for minimization you use
the CPU version of the code. Only in situations where you are confident the
structure is reasonable, for example if it was a snapshot
from dynamics, should you use the GPU code (SPFP) for
minimization.
The single GPU version of PMEMD is called
pmemd.cuda
while the multi-GPU version is called pmemd.cuda.MPI.
These are built separately from the standard serial and
parallel installations. Before attempting to build the GPU
versions of PMEMD you should have built and tested at least
the serial version of Amber and preferably the parallel
version as well. This will help ensure that basic issues
relating to standard compilation on your hardware and
operating system do not lead to confusion with GPU related
compilation and testing problems. You should also be
reasonably
familiar with Amber's compilation and test procedures. The
minimum requirements for building the GPU version of PMEMD
are, as of the AMBER 14 release:
NVIDIA Toolkit v5.0, 5.5, 6.0, 6.5 or
7.5. (v6.5 or later required for Maxwell
[GTX970/980/Titan-X] cards)
NVIDIA GPU supporting Hardware
Revision 2.0, 3.0, 3.5 and later.
NVIDIA CUDA Driver v319.60 or
later (v337.09 or later required for GTX-780Ti and GTX-Titan-Black)
MPI library for parallel GPU.
(MVAPICH2 v1.8 or later / MPICH2 v1.4p1 or later
recommended)
It is assumed that you have already correctly installed
and tested CUDA support on your GPU.
Before attempting to build
pmemd.cuda
you should make sure you have correctly installed both the
NVIDIA Toolkit (nvcc compiler) and a CUDA supporting NVIDIA
driver. You may want to try downloading the NVIDIA CUDA SDK
(available from
http://www.nvidia.com/ or included as 'samples' in the
toolkit v5.0 or later) and see if you can build that.
Additionally the environment variable CUDA_HOME should be
set to point to your NVIDIA Toolkit installation and $CUDA_HOME/bin/
should be in your path. At the time of release CUDA 5.0 or
later is required with 5.0, 5.5, 6.0, 6.5 and 7.5 having been
tested and officially supported. Note toolkit version 7.0 is
not recommended due to
performance regression and a bug in the GPU selection mechanism. As such 7.0 is considered unsupported.
The current recommended version is CUDA 7.5.
Building and Testing the Default
SPFP Precision Model
Single GPU (pmemd.cuda)
Assuming you have a working CUDA
installation you can build the single GPU version,
pmemd.cuda, using the
default SPFP precision model as follows:
cd $AMBERHOME
make clean
./configure -cuda gnu (or intel)
make install
Next you can run the tests using the
default GPU (the one with the largest memory) with:
make test.cuda
The majority of these tests should
pass. However, given the parallel nature of GPUs, meaning
the order of operation is not well defined, the limited
precision of the SPFP precision model, and variations in the
random number generator on different GPU hardware, it is not
uncommon for there to be several possible failures. You may
also see some tests, particularly the cellulose test, fail on GPUs
with limited memory. You should inspect the diff file
created in the $AMBERHOME/logs/test_amber_cuda/
directory to manually verify any possible failures.
Differences which occur on only a few lines and are minor in
nature can be safely ignored. Any large differences, or if
you are unsure, should be posted to the Amber mailing list
for comment.
Multiple GPU (pmemd.cuda.MPI)
Once you have built and tested the
serial GPU version you can optionally build the parallel version
(if you have multiple GPUs of the same model). Unlike the
CPU code it is not necessary to build the parallel version
of the GPU code in order to access specific simulation
options (except REMD). Thus you only need to build the
parallel GPU code if you plan to run a single calculation
across multiple GPUs. AMBER 12 had poor multi-GPU scaling
for GPUs later than the Fermi (C2050/2070/2090) generation.
This scaling has been improved significantly in AMBER 14 via
the use of peer to peer communication over the PCI-E bus.
While the underlying communication between GPUs is peer to
peer if supported the code still requires MPI in order to
run.
No special software beyond CUDA 5.0
and an MPI library is required to support peer to peer. For
details on determining which GPUs can operate in peer to peer
mode please see the multi-GPU section of the running on GPUs
section below. The
instructions here assume that you can already successfully build the MPI version of the CPU code. If you cannot, then you should
focus on solving this before you move on to attempting to
build the parallel GPU version.
The parallel GPU version of the code
works using MPI v1 or later.
You can build the multi-GPU code as
follows:
cd $AMBERHOME
make clean
./configure -cuda -mpi gnu (or intel)
make install
Next you can run the tests using GPUs
enumerated sequentially within a node (if you have multiple
nodes or more complex GPU setups within a node then you
should refer to the discussion below on running on multiple
GPUs):
export DO_PARALLEL='mpirun -np
2' # for bash/sh
setenv DO_PARALLEL 'mpirun -np 2' # for csh/tcsh
./test_amber_cuda_parallel.sh
Building non-standard Precision Models
You can build different precision
models as described below. However, be aware that this is
meant largely as a debugging and testing issue and NOT for
running production calculations. Please post any
questions or comments you may have regarding this to the
Amber mailing list. You select which precision
model to compile as follows:
cd $AMBERHOME
make clean
./configure -cuda_DPFP gnu
make install
This will produce executables named
pmemd.cuda_XXXX
where XXXX is the precision model selected at configure time
(SPFP or DPFP). You can then test this on the GPU with the
most memory as follows:
cd $AMBERHOME/test/
./test_amber_cuda.sh DPFP (to test the DPFP precision
model)
Testing Alternative GPUs
Should you wish to run the serial GPU tests on a GPU different from the one with the most memory (and lowest
GPU ID if more than one identical GPU exists) then you can
provide this by setting the CUDA_VISIBLE_DEVICES environment
variable. For
example, to test the GPU with ID = 2 and the default SPFP
precision model you would specify:
cd $AMBERHOME
export CUDA_VISIBLE_DEVICES=2
make test.cuda
This will automatically run the
calculation on the GPU with the most memory even if that GPU
is already in use (see below for system settings to have the
code auto select unused GPUs). If you have only a single CUDA capable
GPU in your machine then this is fine, however if you want
to control which GPU is used, for example you have a Tesla
C2050 (3GB) and a Tesla C2070 (6GB) in the same machine and
want to use the C2050 which has less memory, or you want to
run multiple independent simulations using different GPUs
then you manually need to specify the GPU ID to use using the CUDA_VISIBLE_DEVICES environment variable. The environment variable CUDA_VISIBLE_DEVICES lists which devices are visible as a
comma-separated string. For example, if your desktop has two
tesla cards and a Quadro:
In this way you only ever expose a
single GPU to the
pmemd.cuda executable and so
avoid issues with the running of multiple runs on the same GPU. This approach is the basis of how you can control GPU
usage in parallel runs.
If you want to know which GPU a calculation is running on
the value of CUDA_VISIBLE_DEVICES and other GPU specific
information is provided in the mdout file.
To check
whether a GPU is in use or not you can use the nvidia-smi
command. For Tesla series GPUs (K20, K40 etc) this provides
% load information and process information for each GPU. For
GeForce series cards this information is not available via
nvidia-smi so it is better to just check the GPU memory
usage and temperature. You can do this with:
Note running X11 can confuse things a little bit, showing
slightly higher memory usage on one of the GPUs, but it
should still be possible to use this approach to determine
which GPU is in use.
An alternative approach, and the one recommended if you
have multiple GPUs in a single node, and only want to run
single GPU jobs, is to set them
to Persistence and Compute Exclusive Modes. In this mode a GPU
will reject more than one job.
In this situation calculation 1 will run on GPU 0, 2 on
GPU 1, 3 on GPU 2 and 4 on GPU 3. The 5th job will quit
immediately with an error stating that no free GPUs are
available.
cudaMemcpyToSymbol: SetSim copy to cSim failed
all CUDA-capable devices are busy or unavailable
This approach is useful since it means you do not have to
worry about setting CUDA_VISIBLE_DEVICES and you do not have
to worry about accidentally running multiple jobs on the
same GPU. The code
will automatically look for GPUs that are not in use and
will use them automatically and will quit if sufficient free
GPUs are not available. You set Persistence and Compute
Exclusive Modes by running the following as root:
$ nvidia-smi -pm 1
$ nvidia-smi -c 3
The disadvantage of this is that you need to have root
permissions to set it and the setting is also lost on a
reboot. We recommend that you add these settings to your
system's startup scripts. In Redhat or Centos this can be
accomplished with:
Note this approach also works on clusters where queuing
systems understand GPUs as resources and thus can keep track
of total gpus allocated but do not control which GPU you see
on a node.
Multi GPU
The way in which a single calculation runs across multiple
GPUs has been changed in AMBER 14. When originally designed
the PCI-E bus speed was gen 2 x16 and the GPUs were C1060 or
C2050s. Since then we have seen GPUs advance to GTX-Titan-Black
and K40s which are ~ 8x faster than the C1060s (more if we
include that AMBER 14 is >30% faster than AMBER 12) while
the PCI-E bus speed has only increased by 2x (PCI Gen2 x16
to PCI Gen3 x16) and Infiniband interconnects by about the
same amount. This unfortunate situation is all too common in
parallel machines and yet many machine designers do not seem
to appreciate the problems this places on software
designers. The situation is now such that for PME
calculations using traditional MPI it is not possible for
AMBER to scale over multiple GPUs. In AMBER 14 we have
chosen to address this problem by focusing on the use of
Peer to Peer communication within a node. This mode of
operation allows GPUs to communicate directly through the
PCI-E bus without going through the CPU chipset which adds
too much latency. Assuming all your GPUs have PCI-E gen 3
x16 bandwidth and support non-blocking peer to peer
communication it is possible to scale to 4, and with the
next version of AMBER hopefully
8 GPUs within a single node. Determining which GPUs can
communicate by peer to peer is discussed below.
It should be noted that while the legacy MPI and GPU-Direct
methods of multi-GPU communication are still supported, and
will be used by the code automatically if peer to peer
communication is not available, you are very unlikely to see
any speedup by using multiple GPUs for a single job if the
GPUs are newer than C2050s. Multi-node runs are almost
impossible to get to scale. We suggest saving money by
skipping the expensive infiniband interconnect and instead
going for GPU dense nodes. Note the standard 4 x GPU Exxact systems
are designed to support 2 x 2 GPU runs at once using peer to
peer. A recent, but expensive, design that
supports 4 way peer to peer was recently released with
details provided on the
recommended hardware
page.
When running in parallel across multiple
GPUs the selection of which GPU to run on
becomes a little more complicated. We have attempted to
simplify things for you by writing a short CUDA program that
checks which GPUs in your node can support peer to peer
communication. You should download and build this as
follows:
make (assuming you have the
cuda toolkit correctly installed this should build
you the executable gpuP2PCheck.
On most of the Exxact Amber certified machines
this will be pairs of GPUs in the order 0+1 and 2+3.
In other words on a 4 GPU machine you can run a
total of two by two GPU jobs, one on GPUs 0 and 1
and one on GPUs 2 and 3. Running a calculation
across more than 2 GPUs will result in peer to peer
being switched off which will likely mean the
calculation will run slower than if it had been run
on a single GPU. To see which GPUs in your system
can communicate via peer to peer you can run the
'gpuP2PCheck' program you built above. This reports
which GPUs can talk to each other. For example from
an Exxact Quantum TXR411-064R 4xGPU system:
CUDA_VISIBLE_DEVICES is unset.
CUDA-capable device count: 4
GPU0 "GeForce GTX TITAN"
GPU1 "GeForce GTX TITAN"
GPU2 "GeForce GTX TITAN"
GPU3 "GeForce GTX TITAN"
Two way peer access between:
GPU0 and GPU1: YES
GPU0 and GPU2: NO
GPU0 and GPU3: NO
GPU1 and GPU2: NO
GPU1 and GPU3: NO
GPU2 and GPU3: YES
So in this case GPUs 0 and 1 can talk to each
other and GPUs 2 and 3 can talk to each other. Thus
to run a 2 GPU job on GPUs 0 and 1 you would run
with:
If peer to peer communication is working you will see the
following reported in your mdout file.
|---------------- GPU PEER TO PEER INFO -----------------
|
| Peer to Peer support: ENABLED
|
|--------------------------------------------------------
Since peer to peer communication does not involve the CPU
chipset so it is possible, unlike in previous versions of
AMBER, to run multiple multi-GPU runs on a single node. In
our 4 GPU example above we saw that GPUs 0 and 1 can
communicate via peer to peer and GPUs 2 and 3 can
communicate. Thus we can run the following options without
the jobs interfering with each other performance wise:
Option 1: All single GPU
export CUDA_VISIBLE_DEVICES=0
cd run1
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
export CUDA_VISIBLE_DEVICES=1
cd run2
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
export CUDA_VISIBLE_DEVICES=2
cd run3
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
export CUDA_VISIBLE_DEVICES=3
cd run4
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
Option 2: One dual GPU and 2 single GPU runs
export CUDA_VISIBLE_DEVICES=0,1
cd run1
nohup mpirun -np 2 $AMBERHOME/bin/pmemd.cuda.MPI
-O -i ... &
export CUDA_VISIBLE_DEVICES=2
cd run2
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
export CUDA_VISIBLE_DEVICES=3
cd run3
nohup $AMBERHOME/bin/pmemd.cuda -O -i ... &
Option 3: Two dual GPU runs
export CUDA_VISIBLE_DEVICES=0,1
cd run1
nohup mpirun -np 2 $AMBERHOME/bin/pmemd.cuda.MPI
-O -i ... &
There are a number of considerations
above and beyond those typically used on a CPU for
maximizing the performance achievable for a GPU accelerated
PMEMD simulation. The following provides some tips for
ensuring good performance.
Avoid using small values of NTPR,
NTWX, NTWV, NTWE and NTWR. Writing to the output,
restart and trajectory files too often can hurt
performance even on CPU runs, however, this is more
acute for GPU accelerated simulations because there is a
substantial cost in copying data to and from the GPU.
Performance is maximized when CPU to GPU memory
synchronizations are minimized. This is achieved by
computing as much as possible on the GPU and only
copying back to CPU memory when absolutely necessary.
There is an additional overhead in that performance is
boosted by only calculating the energies when absolutely
necessary, hence setting NTPR or NTWE to low values will
result in excessive energy calculations. You should not
set any of these values to less than 100 (except 0 to
disable them) and ideally use values of 500 or more.
>100000 for NTWR is ideal.
Avoid setting ntave /= 0. Turning
on the printing of running averages results in the code
means needing to calculate both energy and forces on every
step. This can lead to a performance loss of 8% or more
when running on the GPU. This can also affect
performance on CPU runs although the difference is not
as marked. Similar arguments apply to setting the value
of ene_avg_sampling to small values.
Avoid using the NPT ensemble (NTB=2)
when it is not required; if needed make use of the Monte
Carlo barostat (barostat=2). Performance will generally be NVE>NVT>NPT.
(NVT ~ NPT for barostat=2).
Avoid the use of GBSA in implicit solvent GB
simulations unless required. The GBSA term is calculated
on the CPU and thus requires a synchronization between
GPU and CPU memory on every MD step as opposed to every
NTPR or NTWX steps when running without this option.
Use the Berendsen Thermostat (ntt=1)
or Anderson Thermostat (ntt=2) instead of the Langevin
Thermostat (ntt=3). Langevin simulations require very
large numbers of random numbers which slows performance
slightly.
Do not assume that for small
systems the GPU will always be faster. Typically for GB
simulations of less than 150 atoms and PME simulations
of less than 9,000 atoms it is not uncommon for the CPU
version of the code to outperform the GPU version on a
single node. Typically the performance differential
between GPU and CPU runs will increase as atom count
increases. Additionally the larger the non-bond cutoff
used the better the GPU to CPU performance gain will be.
When running in parallel across
multiple GPUs you should NOT attempt to share nodes and
thus interconnects. For example you should avoid running
2 separate MPI jobs on individual nodes. For example if
you have 2 nodes, each with a QDR IB card in, 1 C2050
and 1 C1060 you will likely get very poor performance if
you attempt to run a dual GPU job on the 2 C2050's and a
second dual GPU job on the 2 C1060's. It is also not
advisable to mix GPU models when running in parallel. In
this situation you are advised to physically place both
C2050's in one node and both C1060's in the other. You
could of course run a dual C2050 job across the two
nodes and then 2 single GPU jobs on each of the C1060's.
It should be noted that for more modern GPUs such as K40
and GTX-780 it is recommended to only run multi-GPU
calculations in the peer to peer intranode mode.
Infiniband is simply not fast enough to cope with modern
GPUs and so it is more efficient to save money on the
interconnect and have multi-GPU nodes.
Turn off ECC (C2050 and later). ECC can cost you up to 10% in performance and hurts
parallel scaling. You should
verify that your GPUs are working correctly, and not
giving ECC errors for example before attempting this. You can turn
this off on Fermi based cards and later by running the
following command for each GPU ID as root, followed by a
reboot:
nvidia-smi -g 0 --ecc-config=0
(repeat with -g x for each
GPU ID)
Extensive testing of AMBER on a wide range of
hardware has established that ECC has little to no
benefit on the reliability of AMBER simulations. This is
part of the reason it is acceptable (see
recommended hardware)
to use the GeForce gaming cards for AMBER simulations.
Turn on boost clocks if supported. Newer GPUs from
NVIDIA, such as the K40, support boost clocks which
allow the clock speed to be increased if there is power
and temperature headroom. This must be turned on to
obtain optimum performance with AMBER. If you have a K40
or newer GPU supporting boost clocks then run the
following:
K40:
sudo nvidia-smi -i 0 -ac 3004,875
which puts device 0 into the highest boost state.
K80:
sudo
nvidia-smi -i 0 -ac 2505,875
To return to normal do:
sudo nvidia-smi -rac
To enable this setting without being root do:
nvidia-smi -acp 0
If you see that performance when running multiple - multi-GPU runs is bad.
That is that say you run 2 x 2GPU jobs and they don't
both run at full speed as if the other job was never
running then make sure
you turn off thread affinity within your MPI implementation or at least set each MPI thread
to use a difference core. In my experience MPICH
does not have this on by default
and so no special settings are needed however both
MVAPICH and OpenMPI set thread affinity by
default. This would actually be useful if they did it in
an intelligent way. However, it seems they pay no
attention to load or even other MVAPICH or OpenMPI runs
and always just assign from core 0. So 2 x 2 GPU jobs
are, rather foolishly, assigned to cores 0 and 1 in both
cases. The simplest solution here is to just disable
thread affinity as followins:
For examples of hardware that has been optimized for
running GPU AMBER simulations, in terms of both price and
performance please see the
following page.