AMBER 14 NVIDIA GPU
ACCELERATION SUPPORT
Benchmarks
Benchmarks timings by Ross
Walker.
This page provides benchmarks for AMBER v14 (PMEMD) with
NVIDIA
GPU acceleration. If you are using AMBER v12 please see
the archived AMBER
version 12 benchmarks.
Download AMBER 14
Benchmark Suite
Please Note: The current benchmark
timings here are for the current version of AMBER 14 (GPU support revision
14.0.1., June 2014) and where labeled the Exxact AMBER Certified
optimized version. (14.1.0.Exx, Dec 2015).
The benchmark numbers in the plots below
labeled 'Exxact' are for the performance of the Exxact optimized
version of AMBER 14 that ships with Exxact AMBER Certified systems
ordered after Dec 20th 2015.
Machine Specs
Machine
Exxact AMBER Certified 2U GPU Workstation
CPU = Dual x 8 Core Intel E5-2650v3 (2.3GHz), 64 GB DDR4 Ram
(note the cheaper 6 Core E5-2620v3 CPUs would also give the same
performance for GPU runs)
MPICH2 v1.5 - GNU v4.4.7-3 - Centos 6.5
CUDA Toolkit NVCC v7.5
NVIDIA Driver Linux 64 - 352.63
Code Base = AMBER 14 Update 13 (GPU 14.0.1 Jun 2014)
/ AMBER 14 Update 13 + Exxact Optimizations (GPU 14.1.0.Exx
Dec 2015)
Precision Model = SPFP (GPU), Double Precision
(CPU)
Parallel Notes = All multi-GPU runs are
intranode with GPU pairs that support peer to peer communication. In
the case of the Exxact machine used here this is device IDs 0 & 1 or
2 & 3.
4 x K40 / Titan-X numbers = The 4 x K40
and Titan-X single
run numbers are from unique hardware, developed in
collaboration with
Exxact, employing 2 x 4 way PCI-E gen 3 x16 switches to
support nonblocking 4 and 8 way peer to peer. More details about how to purchase such hardware
is provided on the recommended
hardware page.
K80 and Titan-Z numbers = The K80 and
Titan-Z cards consist of two physical GPUs per board. Thus if you
have a system with 4 K80 cards in it nvidia-smi will report 8 GPUs.
From an AMBER perspective the GPUs are treated independently. Thus
you could run 8 individual single GPU simulations, 4 dual GPU
simulations or 2 quad GPU simulations or any combination in between.
For the purposes of showing benchmarks here we report the
performance for a single GPU (half a K80 board), dual GPU (a full
K80 board) and quad GPU (two full K80 boards in PCI slots attached
to the same CPU and thus capable of P2P communication).
Benchmarks were run with ECC turned OFF - we have seen no issues with
AMBER reliability related to ECC being on or off. If you see approximately 10% less
performance than the numbers here then run the following (for each GPU) as root:
nvidia-smi -g 0
--ecc-config=0 (repeat
with -g x for each GPU ID)
Boost Clocks: Some of the latest NVIDIA
GPUs, such as the K40 and K80, support boost clocks which increase the clock
speed if power and temperature headroom is available. This should be
turned on as follows to enable optimum performance with AMBER:
K40:
sudo nvidia-smi -i 0 -ac 3004,875
K80:
sudo nvidia-smi -i 0 -ac 2505,875
which puts device 0 into the highest boost state.
To return to normal do: sudo
nvidia-smi -rac
To enable this setting without being root do:
nvidia-smi -acp 0
List of Benchmarks
Explicit Solvent (PME)
- DHFR NVE HMR 4fs = 23,558 atoms
-
DHFR NPT HMR 4fs = 23,558 atoms
-
DHFR NVE = 23,558 atoms
- DHFR NPT = 23,558 atoms
- FactorIX NVE = 90,906 atoms
- FactorIX NPT = 90,906 atoms
- Cellulose NVE = 408,609 atoms
- Cellulose NPT = 408,609 atoms
- STMV NPT HMR 4fs = 1,067,095 atoms
Implicit Solvent (GB)
- TRPCage = 304 atoms
- Myoglobin = 2,492 atoms
- Nucleosome = 25,095 atoms
You can download a tar file containing the input
files for all these benchmarks
here
(87.8 MB)
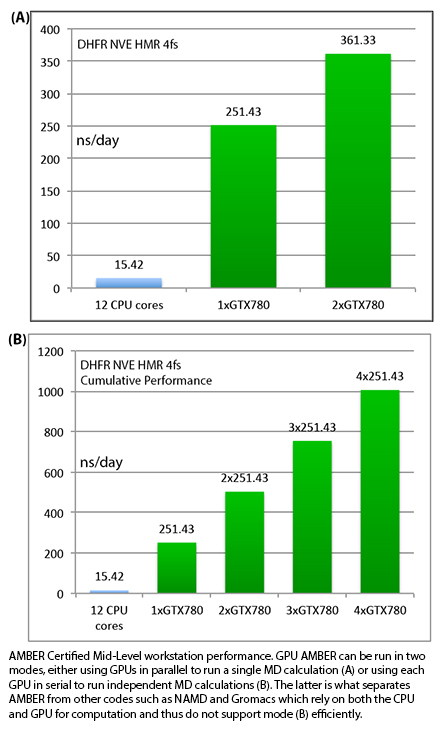
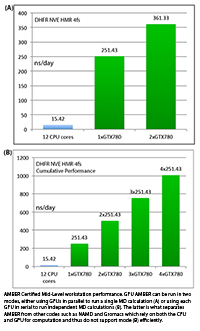
Individual vs Aggregate Performance
A unique feature of AMBER's GPU support that sets it apart from the
likes of Gromacs and NAMD is that it does NOT rely on the CPU to
enhance performance while running on a GPU. This allows one to make
extensive use of all of the GPUs in a multi-GPU node with maximum
efficiency. It also means one can purchase low cost CPUs making GPU
accelerated runs with AMBER substantially more cost effective than
similar runs with other GPU accelerated MD codes.
For example, suppose you have a node with 4
GTX-Titan-X GPUs in it. With a lot of other MD codes you can use one
to four of those GPUs, plus a bunch CPU cores for a single job.
However, the remaining GPUs are not available for additional jobs
without hurting the performance of the first job since the PCI-E bus
and CPU cores are already fully loaded. AMBER is different. During a
single GPU run the CPU and PCI-E bus are barely used. Thus you have
the choice of running a single MD run across multiple GPUs, to
maximize throughput on a single calculation, or alternatively you
could run four completely independent jobs one on each GPU. In this
case each individual run, unlike a lot of other GPU MD codes, will
run at full speed. For this reason AMBER's aggregate throughput on
cost effective multi-GPU nodes massively exceeds that of other codes
that rely on constant CPU to GPU communication. This is illustrated
below in the plots showing 'aggregate' throughput.
^
|


|
|
Explicit Solvent PME Benchmarks |
|
1) DHFR NVE HMR 4fs = 23,558 atoms
Typical Production MD NVE with
GOOD energy conservation, HMR, 4fs.
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2, tol=0.000001,
nstlim=75000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.004, cut=8.,
ntt=0, ntb=1, ntp=0,
ioutfm=1,
/
&ewald
dsum_tol=0.000001,
/
|
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
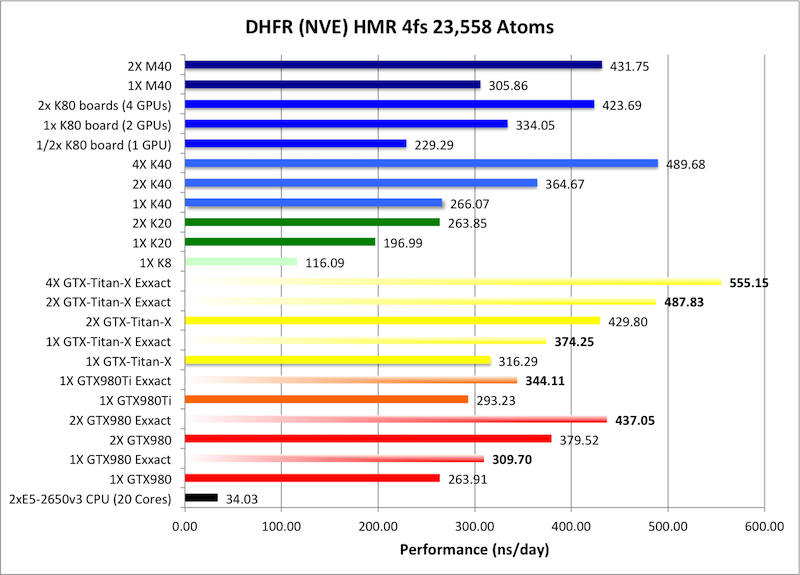
Bold = Exxact Optimized version
of AMBER 14.
|
|
Aggregate throughput (GTX-Titan-X)
(individual runs at the same time on the same node)
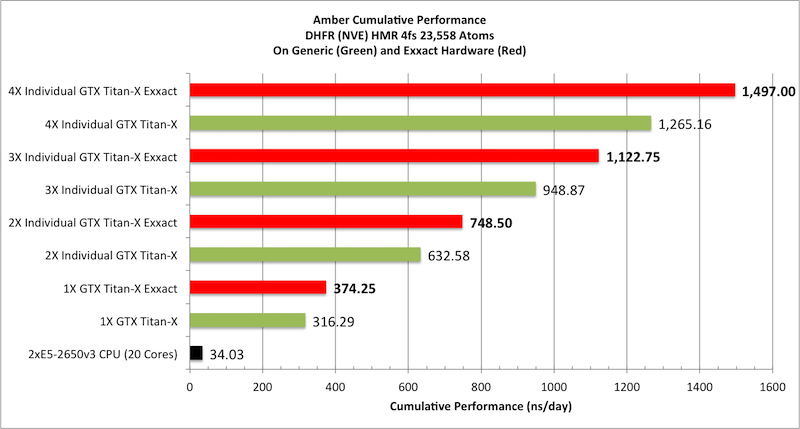
|
|
2) DHFR NPT HMR 4fs = 23,558 atoms
Typical Production MD NPT, MC Bar 4fs HMR
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2,
nstlim=75000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.004, cut=8.,
ntt=1, tautp=10.0,
temp0=300.0,
ntb=2, ntp=1, barostat=2,
ioutfm=1,
/
|
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
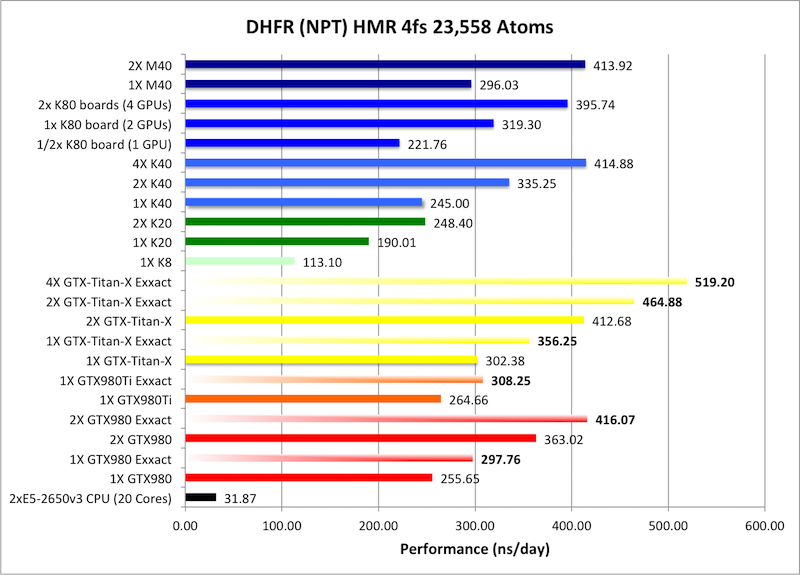
Bold = Exxact Optimized version
of AMBER 14.
|
|
3) DHFR NVE 2fs = 23,558 atoms
Typical Production MD NVE with
good energy conservation, 2fs.
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2, tol=0.000001,
nstlim=75000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=0, ntb=1, ntp=0,
ioutfm=1,
/
&ewald
dsum_tol=0.000001,
/
|
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
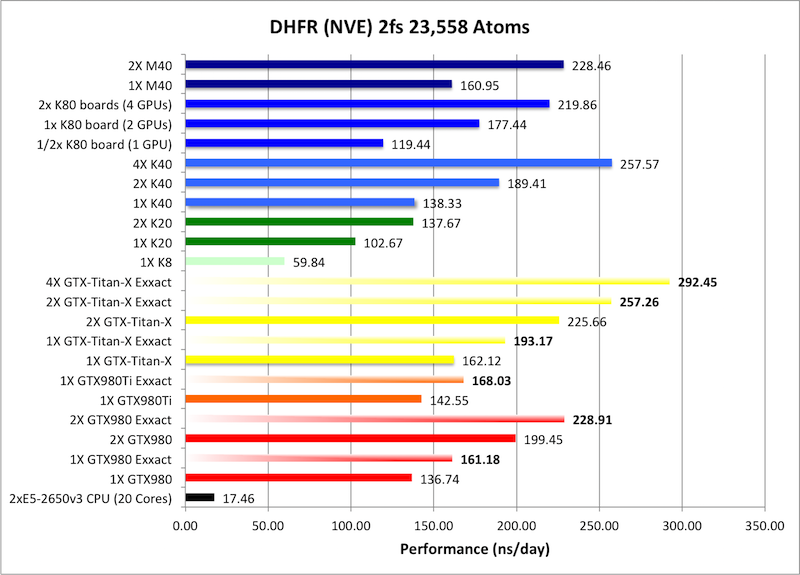
Bold = Exxact Optimized version
of AMBER 14.
|
|
4) DHFR NPT 2fs = 23,558 atoms
Typical Production MD NPT, MC Bar 2fs
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2,
nstlim=75000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=1, tautp=10.0,
temp0=300.0,
ntb=2, ntp=1, barostat=2,
ioutfm=1,
/
|
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
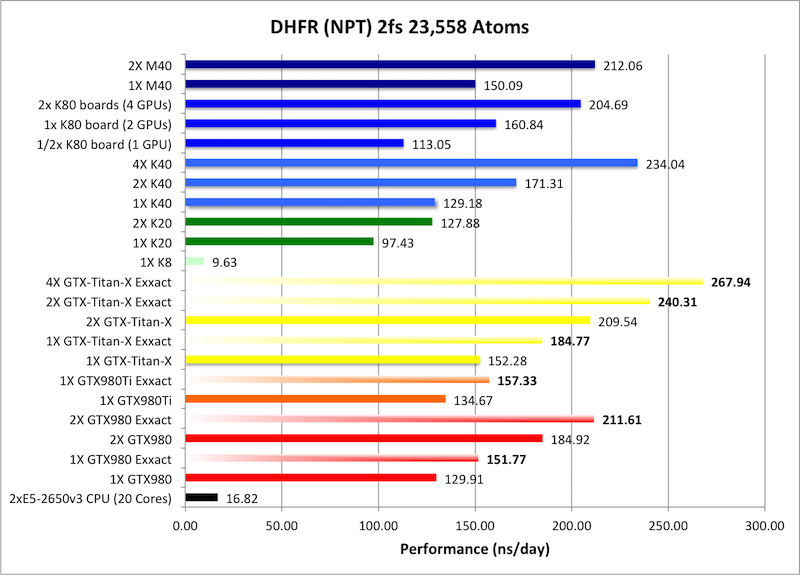
Bold = Exxact Optimized version
of AMBER 14.
|
|
5) Factor IX NVE = 90,906 atoms
Typical Production MD NVE with
GOOD energy conservation.
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2, tol=0.000001,
nstlim=15000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=0, ntb=1, ntp=0,
ioutfm=1,
/
&ewald
dsum_tol=0.000001,nfft1=128,nfft2=64,nfft3=64,
/
|
 |
Single job throughput
(a single run on one or more GPUs within a single node)
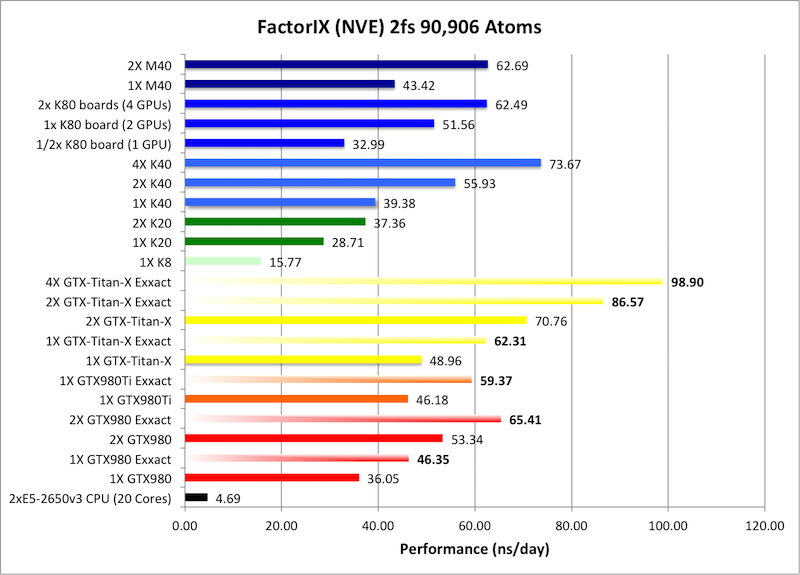
Bold = Exxact Optimized version
of AMBER 14.
|
|
6) Factor IX NPT = 90,906 atoms
Typical Production MD NPT, MC Bar 2fs
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2,
nstlim=15000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=1, tautp=10.0,
temp0=300.0,
ntb=2, ntp=1, barostat=2,
ioutfm=1,
/
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
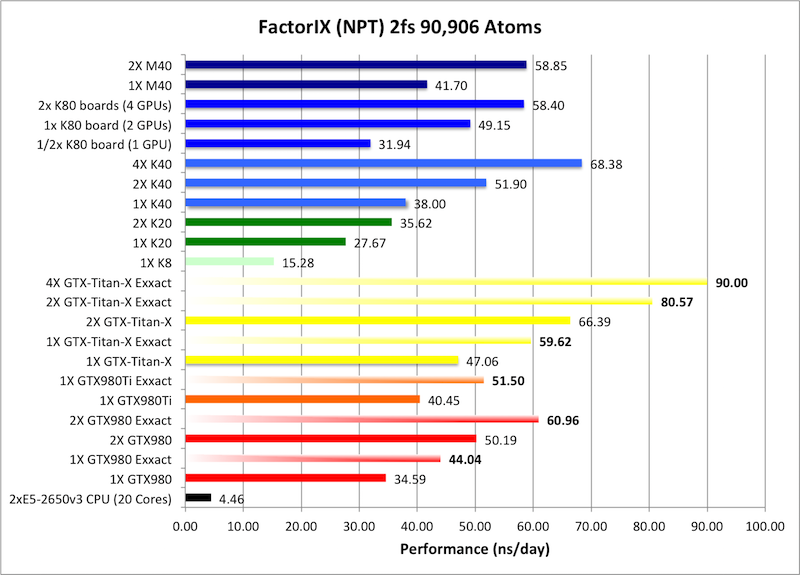
Bold = Exxact Optimized version
of AMBER 14.
|
|
7) Cellulose NVE = 408,609 atoms
Typical Production MD NVE with
GOOD energy conservation.
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2, tol=0.000001,
nstlim=10000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=0, ntb=1, ntp=0,
ioutfm=1,
/
&ewald
dsum_tol=0.000001,
/
|
 |
Single job throughput
(a single run on one or more GPUs within a single node)
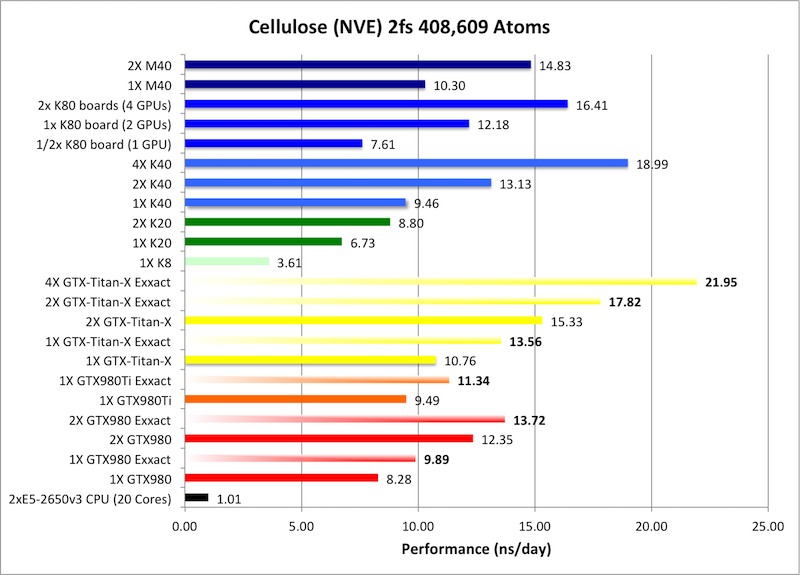
Bold = Exxact Optimized version
of AMBER 14.
|
|
8) Cellulose NPT = 408,609 atoms
Typical Production MD NPT, MC Bar 2fs
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2,
nstlim=10000,
ntpr=1000, ntwx=1000,
ntwr=10000,
dt=0.002, cut=8.,
ntt=1, tautp=10.0,
temp0=300.0,
ntb=2, ntp=1, barostat=2,
ioutfm=1,
/
|
|
Single job throughput
(a single run on one or more GPUs within a single node)
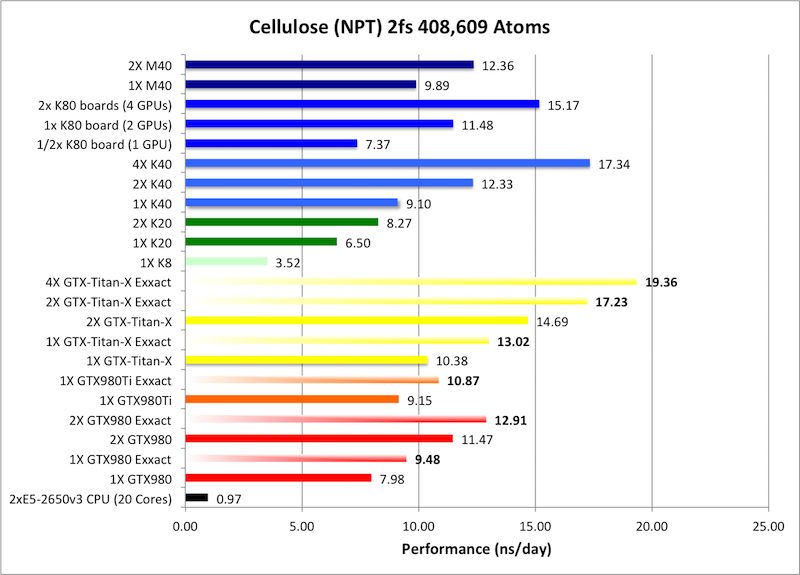
Bold = Exxact Optimized version
of AMBER 14.
|
|
9) STMV NPT HMR 4fs = 1,067,095 atoms
Typical Production MD NPT, HMR, MC Bar 4fs
&cntrl
ntx=5, irest=1,
ntc=2, ntf=2,
nstlim=4000,
ntpr=1000, ntwx=1000,
ntwr=4000,
dt=0.004, cut=8.,
ntt=1, tautp=10.0,
temp0=300.0,
ntb=2, ntp=1, barostat=2,
ioutfm=1,
/
|
 |
Single job throughput
(a single run on one or more GPUs within a single node)
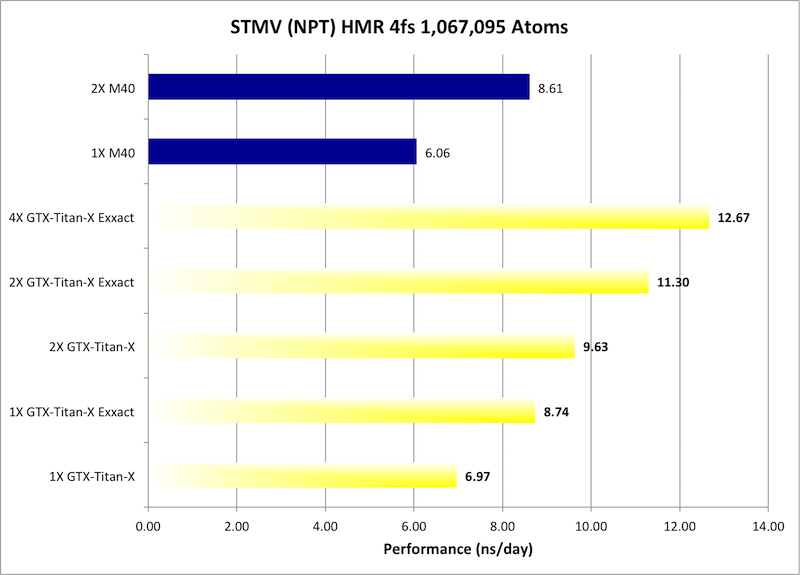
Bold = Exxact Optimized version
of AMBER 14.
|
|
^
|
Implicit Solvent GB Benchmarks |
|
1) TRPCage = 304 atoms
&cntrl
imin=0,irest=1,ntx=5,
nstlim=500000,dt=0.002,
ntf=2,ntc=2,
ntt=1, tautp=0.5,
tempi=325.0, temp0=325.0,
ntpr=1000, ntwx=1000, ntwr=50000,
ntb=0, igb=1,
cut=9999., rgbmax=9999.,
/ Note: The TRPCage test is too small to make effective
use of the very latest GPUs hence
performance on these cards is not as pronounced over early
generation cards as it is for larger GB systems and PME runs.
This system is also too small to run effectively over
multiple GPUs. |
 |
|
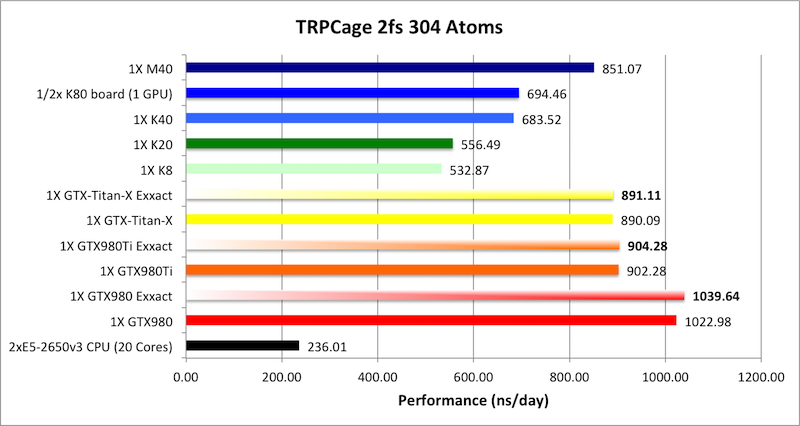
Bold = Exxact Optimized version
of AMBER 14.
TRPCage is too small to effectively scale to multiple modern
GPUs
|
|
2) Myoglobin = 2,492 atoms
&cntrl
imin=0,irest=1,ntx=5,
nstlim=50000,dt=0.002,ntb=0,
ntf=2,ntc=2,
ntpr=1000, ntwx=1000,
ntwr=10000,
cut=9999.0, rgbmax=15.0,
igb=1,ntt=3,gamma_ln=1.0,nscm=0,
temp0=300.0,ig=-1,
/
Note: This test case is too small to make effective
use of multiple GPUs when using the latest hardware. |
 |
|
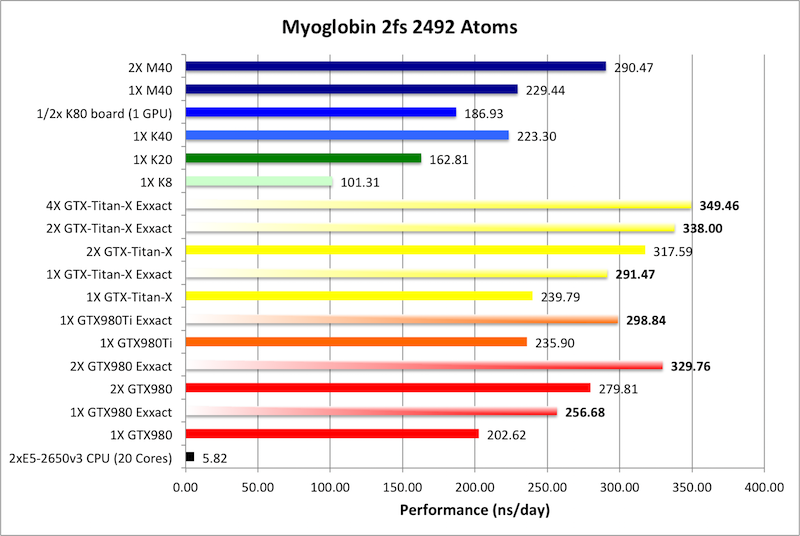
Bold = Exxact Optimized version
of AMBER 14.
Myoglobin is too small to effectively scale to multiple modern
GPUs.
|
|
3) Nucleosome = 25095 atoms
&cntrl
imin=0,irest=1,ntx=5,
nstlim=1000,dt=0.002,
ntf=2,ntc=2,ntb=0,
igb=5,cut=9999.0,rgbmax=15.0,
ntpr=200, ntwx=200,
ntwr=1000,
saltcon=0.1,
temp0=310.0,
ntt=1,tautp=1.0,
nscm=0,
/
|
 |
|
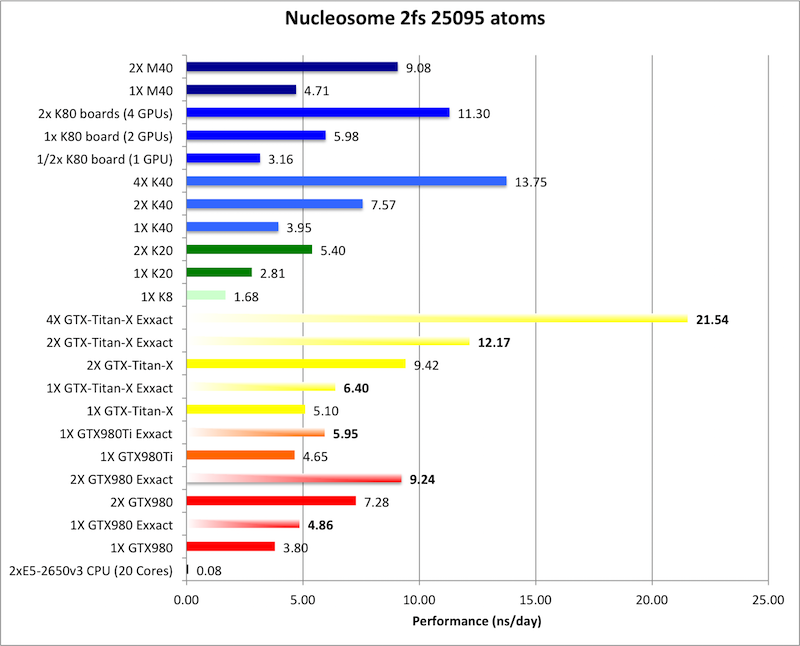
Bold = Exxact Optimized version
of AMBER 14.
|
^
|