Reciprocal Space Convolution of the Long-Ranged Potential
In order to transform the charge density mesh into an electrostatic
potential field, it is necessary to know the interaction potential between any
two mesh points, the way any given mesh point influences one of the others
elsewhere in the box. If the mesh were very fine, it would be precise enough
to simply compute the long-ranged potential
kqiqj erf(αrij) /
rij as if the mesh points were simply particles displaced
from one another by multiples of the grid spacing. However, the mesh would
need to be so fine‐below a tenth of an Å‐that the computation
would be prohibitively expensive. The interpolation done in the
previous section is critical for managing the size of
the mesh problem, and it carries consequences for the form of the potential on
the mesh that must be reckoned with.
Because the convolution is occurring with the help of Fourier transforms to
reduce the algorithmic complexity from O(N2) to O(N log N), the form
of a B-spline in fourier space is important: the Euler exponential
spline. This involves something called the Gamma function (Γ) and the
calculation for our previous
example can be traced from pmeOrthoConvBC to pre-tabulated
quantities in pmeOrthoTabBC and LoadPrefac to the
lowest-level arithmetic in GammaSum, all of which is taken from
the original
Essmann paper.
As one might expect, there is a great deal of pre-computation that can occur
when the simulation box is perfectly rectangular‐it gets tougher when the
box doesn't have all right angles, but the mdgx program in
AmberTools finds additional ways to pre-compute
some of the quantities.
One important thing about Particle-Mesh Ewald calculations which can be seen
in pmeOrthoConvBC is the zeroing of the first reciprocal space
element, that is index (1, 1, 1) of the newly transformed charge density grid.
For net neutral systems, this number will already be zero, give or take minute
amounts depending on the floating point precision, but for systems with a net
charge, the long-ranged effect of the net charge will thus be removed. (We
still recommend that biomolecular simulations be charge neutral.)
There is a lot of dense math. Delving deeper, one discovers things like
"the convolution of a Gaussian is another Gaussian" and other mathematical
relationships that have been used to great effect in variations on the central
Particle-Particle Particle-Mesh technique. It's helpful to visualize the
long-ranged pair potential by which one mesh point influences another, however,
and this can be done with minimal effort following a procedure mentioned in
the Introduction.
Q = zeros(64, 64, 64); % Empty charge grid
Q(32,32,32) = 332.0636; % Proton charge scaled to yield final
% results in kcal/mol
gdim = [64 64 64]; % Grid dimensions
S = 0.5/getalpha(8.0, 1.0e-5); % Gaussian charge spreading factor
ordr = [4 4 4]; % Fourth-order interpolation everywhere
U = pmeOrthoConvBC(Q, gdim, ordr, S);
A slice of this result, the electrostatic potential due to a single mesh
grid point occupied by a charge of +1 proton units, shows in detail the
interaction of any two mesh points. It can be used as a lookup table
indexed relative to the (32,32,32) charge index: for the interaction of mesh
points located at (5,5,5) and (8,10,12), look up the potential in U at
(32+8-5 = 35, 32+10-5=37, 32+12-5=39) and multiply by the charges (proton
units) at each of the mesh points.
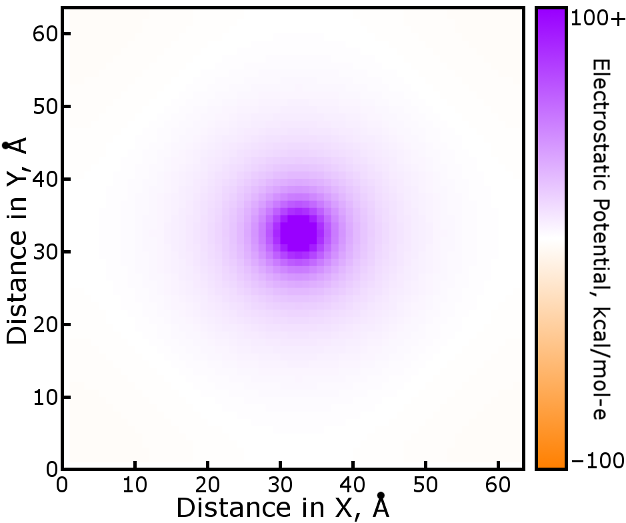
As would be expected, the shape of the potential is roughly circular
(spherical in three dimensions), but not quite: if the box were much
smaller it would become apparent in the image. The potential is a lot like
kqiqj erf(αrij) /
rij, but not quite: the nature of the B-splines
in Fourier space can be examined by returning additional arguments from
pmeOrthoConvBC. Of course, nowhere is the pair potential
negative, but individual charges can be and will scale it accordingly.
The code should make clear that this pair potential is an analytic result
obtained for a particular box size and mesh dimensions. The result U could be
stored, pre-transformed by 3DFFT even, for inclusion in the convolution of any
charge mesh with the same dimensions. In fact, this could save some time in
the reciprocal space calculation but it's trivial in the grand scheme of
things. The analytic approach is nice because, if the simulation box size
changes, the pair potential will automatically update.
The pair potential above is the key to understanding why PME, even with the
FFT, is a fundamentally pairwise operation. The FFT merely makes it a lot
faster for dense systems of particles. For the 4th order
interpolation in this example, the 64-element stencils projecting each of two
charges onto the charge grid would interact according to this potential (even
if those stencils overlap‐note that the potential is not infinite even at
(32,32,32)), implying 4096 table lookups to compute the long-ranged component
of the particles' interaction in a pairwise sense.
An understanding of this material may make it more comfortable to get into
the &ewald namelist of sander or pmemd job
control files. There are still a number of algorithmic constraints that limit
the options that can take effect in the workhorse, GPU version of
pmemd (the grid size must be a multiple of 4, for example), but
that may improve with time. More broadly, this demonstration aims to teach
the methods behind particle-particle, particle-mesh techniques, which have
applications throughout computational physics. Any questions or comments
may be forwarded to david.cerutti (at) rutgers.edu.
Go to the previous section
|


