


Constructing the unit cell: Where crystal simulations depart from ordinary MD
A crystal unit cell is composed of a number of copies of the crystallographic asymmetric unit, which move independently of one another but whose average positions are nonetheless related by the symmetry operations of the space group. Protein X-ray structures found in the PDB will list the positions of atoms in one asymmetric unit and (as described in the previous section) provide the transformations necessary to complete the unit cell. Simulations of protein or nucleic acid crystals are, in principle, no diffferent than simulations of other periodic condensed phase systems; in these simulations, the simulation cell typically corresponds to one unit cell, although in principle the simulation cell can contain multiple unit cells.
In order to construct the unit cell, the PDB file may first need some modification. Open the file 1AHO.pdb in the XtalUtilities directory. This is a high-resolution structure of a scorpion toxin protein; diffraction was performed at 287K and the crystallization conditions were very mild. By all standards we have discussed thus far, molecular simulations of this system are very feasible. However, there are still some details of the PDB file that must be addressed to make the setup proceed smoothly.
First, a number of residues are observed in multiple conformations (Asp9, Cys12, Cys63, and Glu24). The occupancies of each state suggest that they are all equally populated. In some cases, it is necessary to model some parts of alternating asymmetric units in different conformations to avoid clashes. However, that is not the case in this structure, and for simplicity all residues should be modeled solely in the "A" state. Modify the PDB file to eliminate the "B" state in each of these residues. For example,
becomes, after modeling only the "A" state:ATOM 126 CG AASP A 9 10.198 -9.003 0.070 0.50 15.80 C ATOM 127 CG BASP A 9 7.851 -9.100 1.119 0.50 16.40 C ATOM 128 OD1AASP A 9 10.052 -8.373 -1.046 0.50 16.38 O ATOM 129 OD1BASP A 9 7.259 -9.001 -0.022 0.50 17.40 O
ATOM 126 CG ASP A 9 10.198 -9.003 0.070 0.50 15.80 C ATOM 128 OD1 ASP A 9 10.052 -8.373 -1.046 0.50 16.38 O
There are also some AMBER-specific modifications to make to the PDB file, to ensure that it is read properly by tLEaP later in the setup. These include relabeling all CYS residues CYX, as they are all involved in disulfide bridges, and relabeling the HIS residues HID, as the delta protonation state optimizes hydrogen bonding between these residues and acceptors on other asymmetric units. All of these changes have been made in the PDB file c1AHO.pdb, also provided in the XtalUtilities directory. Users are, of course, encouraged to make their own changes as they follow the tutorial.
We are now ready to construct the unit cell (in this tutorial, the simulation cell will consist of only one unit cell, although we recommend that in practice at least two unit cells be used to avoid bounday artifacts if the unit cell is as small as this one). The program UnitCell can easily process the PDB file we have modified. Starting in the directory XtalUtilities, execute the command (substituting your own file names if you have chosen to complete the process yourself):
[user]$ ${AMBERHOME}/bin/UnitCell -p c1AHO.pdb -o x1AHO.pdb
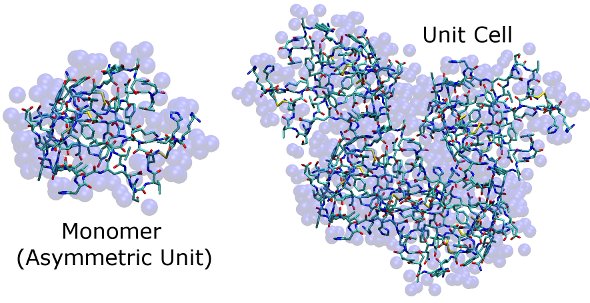
Here are images of the structures in c1AHO.pdb and x1AHO.pdb. The protein is shown in stick form, and water (for which only oxygens are identified) is shown as transparent blue spheres. It is difficult to see how the unit cell fits together at this stage, but so long as the simulation cell dimensions match those found in the CRYST1 record, clashes in our simulation of this high-resolution X-ray structure will be minor and easily removed by a few steps of steepest descent energy minimization.
In order to demonstrate that the unit cell does indeed tile in space, it may be helpful to make use of the program PropPDB. This program replicates the unit cell to produce a "supercell" and is the tool to use in practice when a unit cell is too small to simulate without the risk of introducing boundary artifacts. Execute the command:
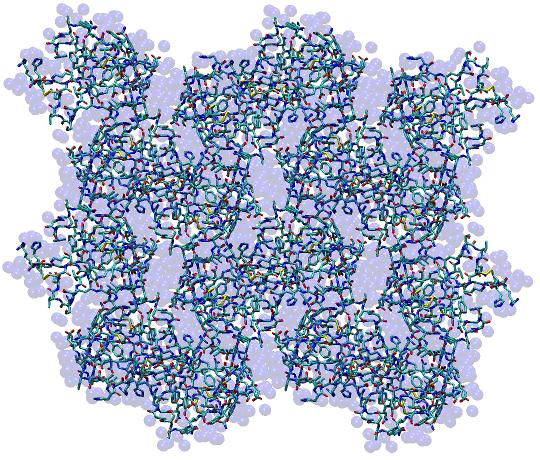
[user]$ ${AMBERHOME}/bin/PropPDB -p x1AHO.pdb -o x8_1AHO.pdb -ix 2 -iy 2 -iz 2
This will create a 2 x 2 x 2 supercell in the PDB file x8_1AHO.pdb (pictured below) which can be visualized with any 3D molecular modeling program to verify that the unit cells do indeed tile in three dimensions.
Continue to the next section.
Return to the beginning of the tutorial.