


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008
Building your own Custom Residues (old version) - SECTION 3
By Ross Walker
Stage 3 - Loading the Protein PDB and Creating a Library File
We are now almost done. We have our non-standard residue defined we now just need to let xleap know about any missing parameters. Before we do this lets set up our protein in xleap with all the necessary bonds, in this way we can have xleap tell us what parameters are missing.
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
In order for xleap to know about our new residue when we load the 1PLC_mod_final.pdb file we need to make sure we load our new library file first. This will define the CUA unit so that when xleap encounters it in the pdb file it will know the topology, charges and atom types:
> loadoff cua.lib
> 1PLC = loadpdb 1PLC_mod_final.pdb
There should now be no error messages. Just the warnings about duplicate atoms.
Now before we go any further we have to ensure that all the bonds to our copper atom are defined. At the moment none of the bonds are defined. We need to add bonds between the copper and the cysteine (84) sulphur atom, between the copper and the MET (92) sulphur atom and between the copper and the delta nitrogen of the two histidines (37 & 87). We can do this with the bond command, alternatively we could edit 1PLC and draw in the bond manually but for a protein this is pretty tedious, try in and you will see what I mean.:
> bond 1PLC.37.ND1 1PLC.100.CU
> bond 1PLC.87.ND1 1PLC.100.CU
> bond 1PLC.84.SG 1PLC.100.CU
> bond 1PLC.92.SD 1PLC.100.CU
Note: the desc command can list the residue number and atoms names so you can identify what to bond. E.g. desc 1PLC.CUA.
This will create the four missing bonds. We can now go ahead and solvate our system in a truncated octahedral box of TIP3P water.
> solvateoct 1PLC TIP3PBOX 12
This will add a 12 angstrom buffer of water around our system. In addition to the already defined crystallographic waters. Next we charge neutralise our system since it currently has a charge of -9.0 (check 1PLC). We shall therefore add a total of 9 Na+ ions.
> addions 1PLC Na+ 9
This may take a few seconds to run. The output should look something like this.
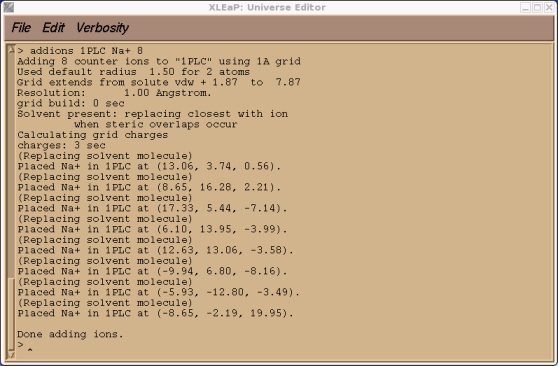
Now we can try and save our prmtop and inpcrd files. At this point we should find that xleap cannot find the type CU.
> saveamberparm 1PLC 1PLC.prmtop 1PLC.inpcrd
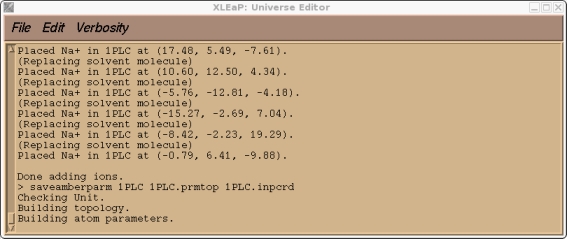
This is to be expected since CU was an atom type that I made up that currently doesn't exist in the standard PARM99 force field. If you check our 1PLC unit you should also find, along with a large number of close contacts due to the solvation, a number of missing parameters all corresponding to the new CU atom type.
> check 1PLC
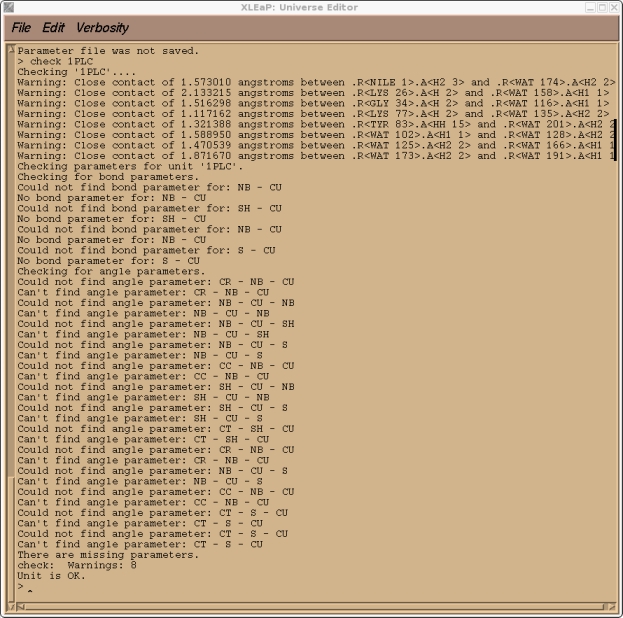
So, before we can go any further we need to add all of these parameters to the AMBER force field. Before we quit xleap however we shall save a 1PLC library file so that we don't have to repeat all of the steps we did above. We can just reload the library file.
> saveoff 1PLC 1PLC.lib
Here it is: 1PLC.lib
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008