


Implementing pairwise 12-6-4 LJ potential using tLEaP
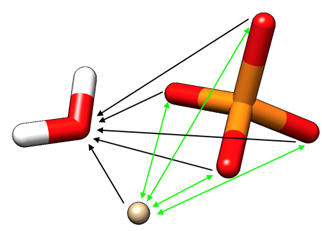
Figure 2.1: A phosphate-water-ion system with solvent C4 (indicated black) and modified C4 (indicated green) terms
Learning Outcomes
Overview
This is a extended tutorial to run an MD simulation using PMEMD.cuda in AMBER24 where metal->water, ligand->water, and metal<->ligand C4 interactions are applied using the novel addC4Pairwise and addC4Type keyword. Please note that this tutorial does not require ParmEd, but will not work for AMBER version prior to (including) AMBER22AmberTools23.
Introduction
In AMBER24's PRMTOP file, the two tLEaP commands (addC4Pairwise and addC4Type) will generate three entries: LENNARD_JONES_CCOEF which was previously generated by ParmEd only, but now can be generated by addC4Type in tLEaP. LENNARD_JONES_DCOEF and LENNARD_JONES_DVALUE that save information for the atom-specific pairwise C4 interaction.
Methods:
Step-by-step explanation:
tleap -s -f leap.in
This requires MOL.frcmod, MOL.mol2, MOL.pdb.sed "s/o o o o /o- o- o- o- /g" resultPO4-nocount5_addc4.prmtop > temp && mv temp resultPO4-nocount5_addc4.prmtop
This is to rename the atom type manually to fulfill criteria #1 (atom type names must have "+" or "-" in them to enable C4).pmemd.cuda -O -i md_min1.in -o md_min1.out -p resultPO4-nocount5_addc4.prmtop -c resultPO4-nocount5.inpcrd -r md_min1.rst -x md_min1.netcdf -inf md_min1.mdinfo -ref resultPO4-nocount5.inpcrd
- The first step of minimization should give a VDW energy of about 5888 kcal/mol.
By Zhen Li