


Copyright (C) Pengfei Li 2016
Setting Up A Protein System at the FCC Metal Surface
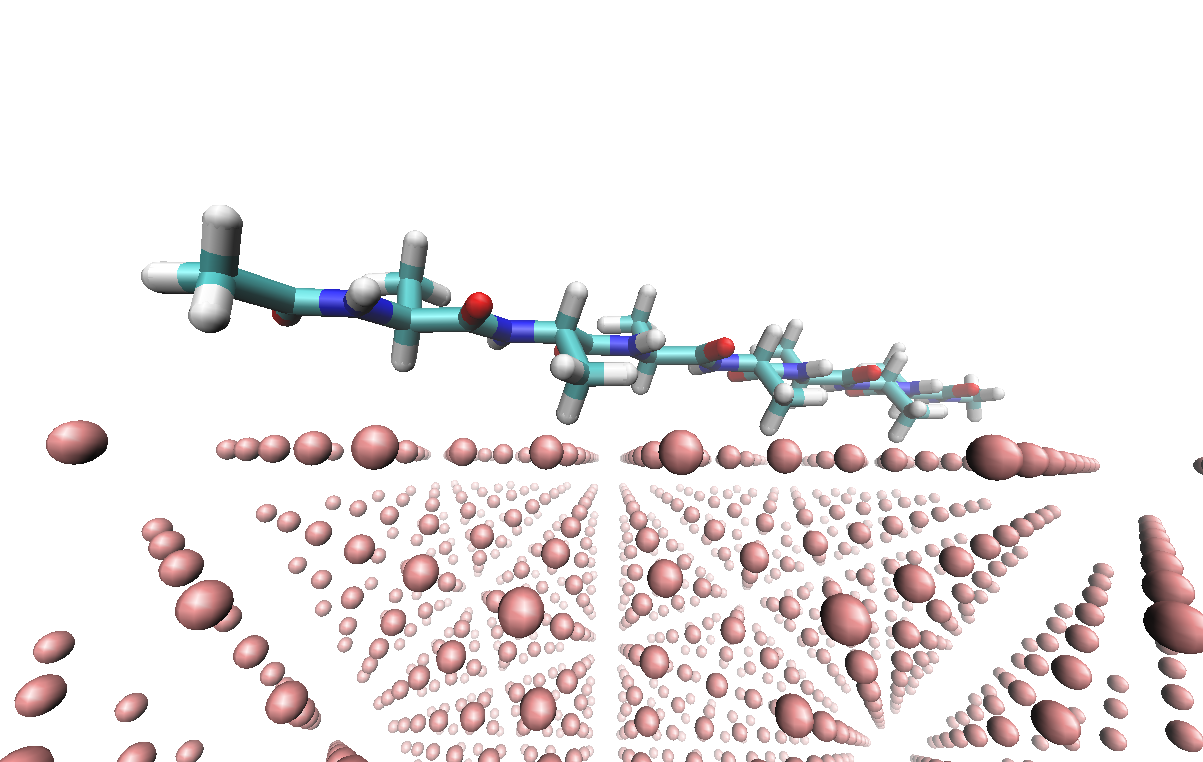
The FCC metal parameters are from Heinz et al.[1] Systematic simulation works have been done for the peptide-metal interaction, one example could be found here.[2]
1. Generate the protein pdb file:
First, we can generate a peptide with sequence of ACE-ALA7-NME using tleap. Here is the input file: seq.in. Then we can perform the command:
tleap -s -f seq.in > seq.out
Afterwards we can get the ace-ala7-nme.pdb file.
2. Generate the FCC metal surface pdb file:
Please download the INTERFACE force field package (version 1.5): INTERFACE-MD. Then uncompress it, and you will find the ag_cell_P1_100.car file under the "INTERFACE_FF_1_5/MODEL_DATABASE/METALS/" directory. This file is a small unit with just four Ag atoms which represents the {100} surface of the FCC Ag metal.
Then you can use the VMD software to convert the ag_cell_P1_100.car file to a PDB file named Ag_Cell_P1_100.pdb, and then manually modified the PDB files with all Ag atom separated into different residues and both the residue name and atom name as "Ag0". This is in order to keep consistent with the AMBER compatible INTERFACE force field. Here is the file after manually modification: Ag_Cell_P1_100.pdb.
Then you can use the PropPDB program in AMBER to generate a bigger FCC metal cluster by duplicate the small Ag unit 8*8*8 times:
PropPDB -p Ag_Cell_P1_100.pdb -o Ag_s100_2048.pdb -ix 8 -iy 8 -iz 8
You will get the Ag_s100_2048.pdb file which has 2048 Ag atoms in it.
3. Combine the protein and FCC metal structures:
We can use the cat command to combine the protein and FCC metal PDB files and then use the pdb4amber program to resequence it.
cat ace-ala7-nme.pdb Ag_s100_2048.pdb > pro_metal.pdb
pdb4amber -i pro_metal.pdb -o pro_metal_renum.pdb
However, when we check the structure pro_metal_renum.pdb we wil find the protein and metal surface are not aligned well with each, you can use the VMD program to move the protein (chose "Mouse"->"Move"->"Fragment" and move the protein by mouse) and then save the structure into a new pdb file: pro_metal_adjust.pdb
4. Generate the AMBER topology and coordinate files:
After that, we can use the pro_metal_tleap.in file herein (in which we use the ff14SB for protein[3]) to perform the following command:
tleap -s -f pro_metal_tleap.in > pro_metal_tleap.out
Then we will have the pro_metal.prmtop and pro_metal.inpcrd files for minimization and MD simulations.
5. Perform minimization and MD simulations:
Herein we will perform the minimization and MD simulations under gas phase, therefore we need to use sander or sander.MPI to perform these modelings (pmemd and pmemd.cuda are usually only used for condensed phase simulations). In order to prevent the Ag cluster fall into pieces, during these modelings we will restrained the positions of the Ag cluster by the harmonic potentials. To just show an example, in general herein we only performed three steps: minimization, heating, and production. However, practical research may need much more steps to get meaningful data.
Here are the three input files for different stages: pro_metal_min.in, pro_metal_heat.in, and pro_metal_prod.in
And then we can perform the following commands sequentially to perform these simulations:
sander -O -i pro_metal_min.in -o pro_metal_min.out -p pro_metal.prmtop -c pro_metal.inpcrd -r pro_metal_min.rst -ref pro_metal.inpcrd
sander -O -i pro_metal_heat.in -o pro_metal_heat.out -p pro_metal.prmtop -c pro_metal_min.rst -r pro_metal_heat.rst -x heat.netcdf -ref pro_metal_min.rst
sander -O -i pro_metal_prod.in -o pro_metal_prod.out -p pro_metal.prmtop -c pro_metal_heat.rst -r pro_metal_prod.rst -x prod.netcdf -ref pro_metal_heat.rst
Click here to go back to the main page
References:
[1] Hendrik Heinz, Barry L. Farmer, Rajesh R. Naik, "Accurate simulation of surfaces and interfaces of face-centered cubic metals using 12-6 and 9-6 Lennard-Jones potentials", The Journal of Physical Chemistry C, 2008, 112, pp. 17281-17290
[2] Hendrik Heinz, Barry L. Farmer, Ras B. Pandey, Joseph M. Slocik, Soumya S. Patnaik, Ruth Pachter, Rajesh R. Naik, "Nature of molecular interactions of peptides with gold, palladium, and Pd-Au bimetal surfaces in aqueous solution", Journal of The American Chemical Society, 2009, 131, pp. 9704-9714
[3] James A. Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E. Hauser, Carlos Simmerling "ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB", Journal of The American Chemical Society, 2015, 11, pp. 3696-3713