


Section 1: Prepare the system and set up the restraints
The current APR scripts were written in Python language. To execute them, you need Python version 2.7 installed on your machine. If you don't have access to Python 2.7, consider using the miniconda distribution that is optionally installed in Amber 16. You can invoke this Python version with $AMBERHOME/miniconda/bin/python. Another alternative is to install Python 2.7 through Anaconda (https://www.python.org/download/releases/2.7/).
The scripts will automatically set up the APR framework: building each umbrella sampling window, pulling the guest away from the host, and imposing the necessary restraints for the system along the way. Equilibration, simulation, and data analysis are also carried out in a highly-automated way.
To start the binding calculations, the first thing you need to do is to download apr.tar.gz and extract the files to your local directory:
tar zxvf apr.tar.gz
You should be able to see the following files in your APR folder:
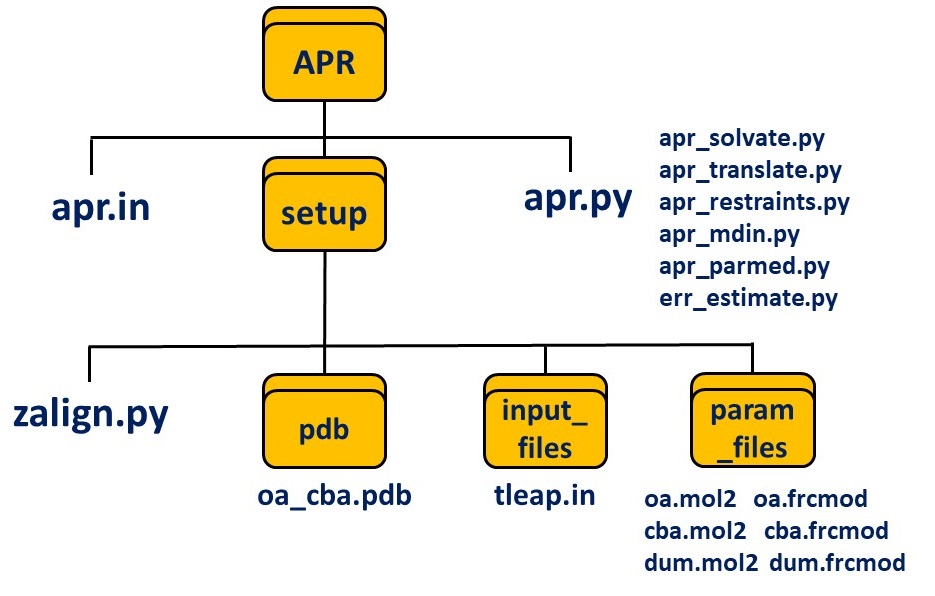
Within the APR folder, issue the command:
(in ./APR)
python2 apr.py
and you will see a greeting message and a brief summary of how to use the scripts.
We will explain the details of files in each subfolder soon. For now, load the oa_cba.pdb file contained in the "pdb" folder (./APR/setup/pdb) in VMD and get familiar with the initial bound structure of OA with the guest. A tutorial of VMD is also available on the Amber tutorial website (http://ambermd.org/tutorials/basic/tutorial2/). The host molecule called octa acid has a bowl-shape cavity and the narrower end of the cavity is essentially closed. Given that the experimental binding measurement was conducted at pH value of ~11, both OA and its guest were modeled as fully deprotonated.
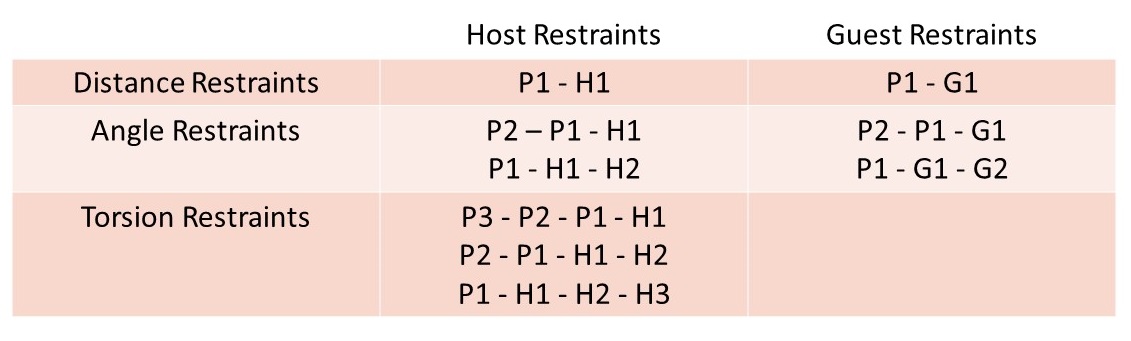
Imposing restraints is crucial for performing binding calculations with the APR method. For the host molecule, distance, angle, and dihedral restraints are used to fix its position and orientation. Note that those restraints do not perturb the internal conformational degrees of freedom of the host, and will stay constant in all umbrella sampling windows. Therefore, there is no need to compute the work of attaching or detaching them.
For the guest, one distance restraint is used to determine how far the guest is pulled away, and two angle restraints control the orientation of the guests. The work of releasing guest restraints is computed analytically. In some cases, it may be necessary to use conformational restraints to increase the distances of pairs of atoms across the rim of the host cavity, so that the guest can overcome the large energy barrier when leaving the host cavity. The addition of the conformational restraints, also called jacks, are not needed in this tutorial example but can be enabled by the APR scripts when large pulling barrier occurs.
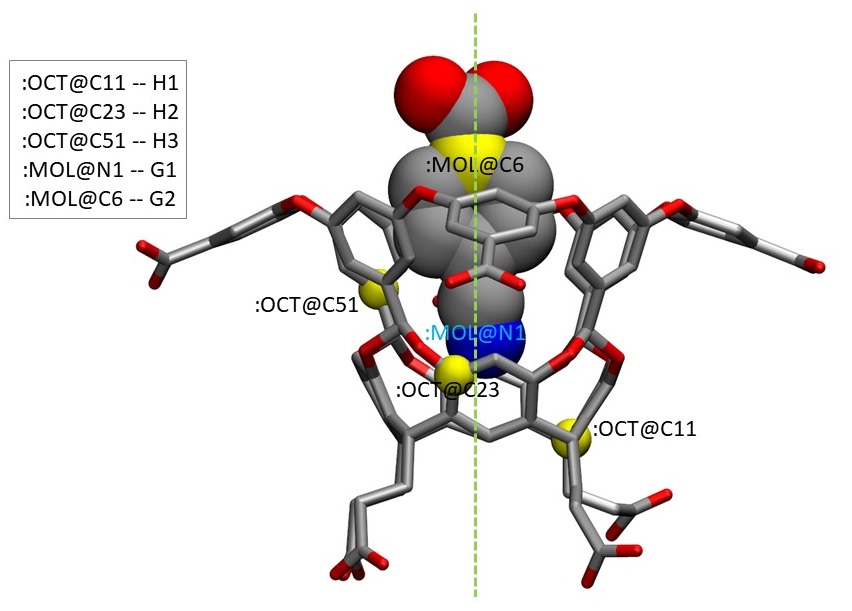
To set up restraints, we need to pick five atoms: three atoms on the host and two atoms on the guest. For the host, we will choose heavy atom C11, C23 and C51 following two general rules: (1) they do not lie in the same line, and (2) they are scattered through the relatively rigid regions of the host. For the guest, we will pick up heavy atom N1 and C6 which are both close to the central axis of the system in the bound state, to avoid clashes with the host molecule during pulling process as much as possible. (Initial structure was obtained from docking and then preliminary MD simulations.) Those five atoms correspond to H1, H2, H3, G1 and G2 in the APR input file apr.in, respectively. H stands for host and G denotes guest. G1 is supposed to be closer to the bottom of the binding cavity than G2, due to the way the restraints are set between dummy atoms and the ligand. OCT and MOL are residue names for the host and the guest in the PDB file, respectively. The Amber mask syntax (":" for residue selection and "@" for atom selection) will be used to indicate those atoms in the following tutorial.
Next, we will use a script called zalign.py to align the central axis of the system with the longest axis (Z) of the rectangular simulation box. Because we want to pull the guest out in a nearly straight line, aligning the host-guest pair with the Z-axis will make things a lot easier. You can now run zalign.py in the "./APR/setup" directory to reorient the host-guest system, by issuing the command
(in ./APR/setup)
python2 zalign.py -f pdb/oa_cba.pdb -a1 :OCT@O3 -a2 :OCT@O7 -a3 :OCT@O8 -a4 :OCT@O11 -c :MOL@N1 -r 85
A file named align_z.pdb will be generated in the "setup" folder. We will not get into too many details of the zalign.py script because it is not directly relevant to the APR method. But if you are interested, you can issue the command
python2 zalign.py -help
to explore the usage of this script. Note that the three host atoms :OCT@O3, :OCT@O7 and :OCT@O8 are three host atoms that you think should lie in the same XY plane after the Z-axis alignment, and they are not related to H1, H2 and H3 we picked above.
Now you can load align_z.pdb to VMD to confirm that the system is now well aligned with the Z-axis.
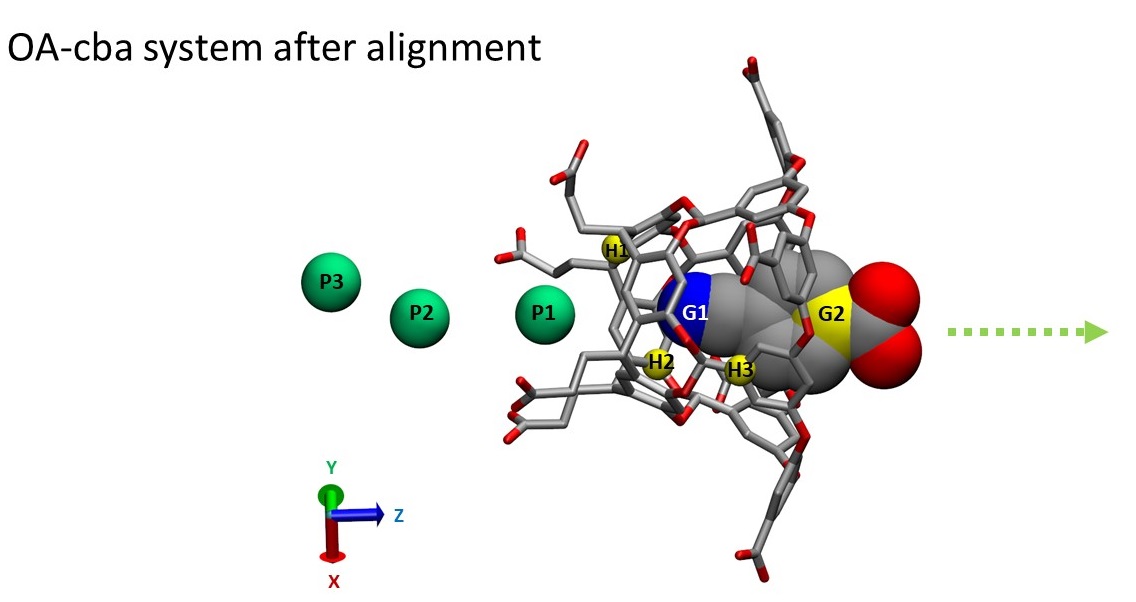
Besides the alignment, zalign.py carries out two more tasks: (1) it sets the coordinates of atom G1 as the new origin, which is specified by -c flag. (2) it appends the entries of three dummy atoms: P1, P2 and P3 to the end of the align_z.pdb file. These dummy atoms were assigned by an atom type "Pb" but they have no charge or volume, yet an atomic mass of 220 Da. The coordinates of the dummy atoms are unchanged during the simulations. Therefore, they can be used as anchor particles to place the following restraints to the system (For more details, please see Henriksen et al. Figure 3 and Figure S1):
In the next section, we will learn how to build APR windows for the attach phase and the pulling phase, and then further look into the details of restraints in each window.