Stage 1: The Molecule in Many, Many Guises
The mdgx &configs module will be called into action once
more, in much the same capacity as at the beginning of the IPolQ tutorial.
This time, we will generate many more configurations of two different molecules
with the aim of sampling a number of degrees of freedom, decorrelating the
motions, and producing inputs ready for quantum calculations. This showcases
the power of the &configs module, and in fact it's just
warming up. In principle, this is nothing you couldn't accomplish with a
shell script writing input files and calling sander, but it would
take half an hour to set up and several minutes to run. After doing that
myself a few hundred times, I wrote the mdgx &configs
module.
The following input will create the conformations we need based on some
initial coordinates for Glycerol using the
topology with vacuum phase charges as
given in the corresponding
.prepi file. As was
stated in the IPolQ tutorial, we will be doing the quantum calculations in
vacuum, so we want to make the conformations and then do the parameter fitting
in the context of vacuum phase charges. We will substitute the IPolQ charges
later. There's a lot of conformations, but don't despair‐these QM
calculations are fast (about 75 seconds each on a 2.6GHz CPU made in 2010).
The conformations sample the major heavy atom dihedral in glycerol and also the
rotation of the central hydroxyl group: both of these motions are sampled
independently thanks to the combine 1 2 command (see the
mdgx &configs keywords glossary by running
$AMBERHOME/bin/mdgx -CONFIGS). While sampling the dihedrals, a
few key angles are pulled towards random values up to 20 degrees away from
their expected equilibria. This may seem pretty savage, but the NMR restraints
that mdgx is using to accomplish this are harmonic potentials that
work against the existing harmonic potentials already present in the topology
created with our initial guess for the force field (in this case GAFF). Only a
few of the angles in the final structures will depart from their standard
configurations by more than 10 degrees. Note that the operations specified by
GridSample, RandomPerturb, and similar keywords get
labeled by numbers in the order they appear so that the combine
(alias LinkOperations) keyword has a handle on them. The user
can also use the label attribute available to each operation to
give them unique and intuitive names for managing very elaborate
manipulations.
&files
-p Glycerol_vac.top
-c Glycerol.crd
-o GenConformers.out
&end
&configs
GridSample :1@O1 :1@C1 :1@C2 :1@O2
{ -180.0 180.0 } Krst 64.0
GridSample :1@C1 :1@C2 :1@O2 :1@HO2
{ -180.0 180.0 } Krst 64.0
RandomSample :1@C2 :1@O2 :1@HO2
{ 90.0 130.0 } Krst 256.0
RandomSample :1@C1 :1@C2 :1@O2
{ 90.0 130.0 } Krst 256.0
RandomSample :1@C1 :1@C2 :1@C3
{ 90.0 130.0 } Krst 256.0
combine 1 2
count 324
verbose 1
% Controls on the quantum mechanical operations
qmlev 'MP2',
basis 'cc-pvDZ',
% Output controls: TAKE NOTE, this will generate a lot of files so
% do this in a clean directory that you won't ls very often.
outbase 'Conf', 'Conf'
write 'pdb', 'orca'
outsuff 'pdb', 'orca'
&end
The point of generating so many conformations is not to make this tutorial
excessively long: rather, it is to emphasize that one must aggressively sample
the conformations of a molecule to ensure that a set of conjured parameters
is correctly calibrated. A cross-section of the conformations this job file
will create is show in the figure below.
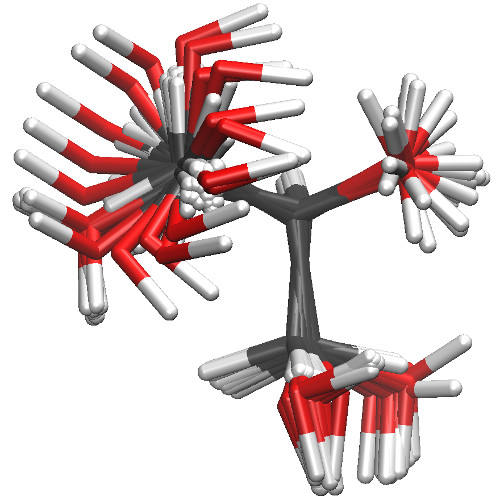
If you have six processors to devote to the problem, a simple shell script
will take care of them all while you go to lunch. For harder systems and more
accurate calculations, it will be necessary to call upon cluster resources and
batch submissions, but this is the nature of the problem. Hopefully,
mdgx can take care of the work of creating the quantum input
files. As the shell script below shows, however, the complete quantum outputs
should be saved in order to take advantage of another new feature of
mdgx.
#!/bin/bash
NJOB=0
for I in `seq 1 324` ; do
<ORCA executable> Conf${I}.orca > Conf${I}.oout &
let "NJOB+=1"
if [ ${NJOB} -eq 6 ] ; then
NJOB=0
wait
fi
done
For those that do not want to crunch the numbers on their own,
here are the glycerol conformations
and here are their ORCA
outputs. Beware: lots of files! Your results may differ slightly as the
initial coordinates I've provided weren't the same ones I took in my own pass
through the tutorial, but there should be no trouble getting a good spread of
conformations.
I've said that parameters do not exist in isolation, but there's more: most
parameters in Amber are shared between more than one molecule because they are
bound to atom types with broader application. One cannot simply retune
parameters for one case and expect them to still serve their original purpose.
However, mdgx makes it very simple to optimize parameters for
multiple systems at once. The ¶m module we are about to
employ was written with the expectation of an ever-expanding data set, and it
has been used to fit nearly a thousand parameters for several hundred
combinations of amino acids simultaneously, involving more than a quarter
million data points.
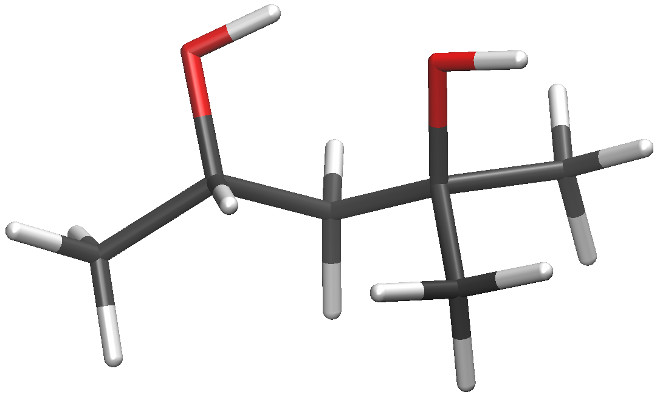
We'll also fit parameters for the larger molecule methyl(2,4)-pentanediol
(MPD) in this exercise. As shown in the figure below, MPD is has more
hydrophobic character than glycerol, but the GAFF atom typing is the same for
both molecules and so they share many bonded parameters. I made some IPolQ
charges for that molecule as well: here are
seed coordinates, its
topology with vacuum phase charges, and the
.prepi file that produced it.
We can use those files, along with a sensible adaptation of the conformational
search job file for glycerol, to sample conformations of MPD. Here is the
input file, here is a
tarball of the conformations, and here
is a tarball of the ORCA
outputs. These calculations each took about 7.5 minutes on our 2.6GHz CPU,
about six times the cost of the glycerol conformations but still pretty
short.
With all of the calculations done, we now have to extract the energies. One
could, of course, just grep for the energies inside each of the
ORCA outputs and write them out next to the Amber coordinates of
the conformations the correspond to. However, this leaves you vulnerable in a
couple of ways: first, in order to script the process successfully, every
calculation needed to work, and in a lot of cases, particularly when trying to
get parameters for drug-sized molecules, the calculations can fail. Worse,
there can be more insidious failures, warnings thrown deep inside the quantum
outputs or matters as simple as a strained configuration obtaining a new
chemical configuration that puts it on a whole new energy surface. A robust
script to read the energies therefore needs to be fault tolerant as well as
programmed to check for problems. This is why mdgx has its own
solution: the &speval module. Given a topology (so that it
knows the number of atoms) and a series of quantum outputs, mdgx
is programmed to check for errors as it extracts coordinates and single point
energies from quantum outputs. (We'll be doing more work under the hood as time
goes on to improve the error checking, but if all goes well you won't notice
anything.) The code you need to run it for the Glycerol case looks like this:
&files
-p Glycerol_vac.top
-o concat.out
-d energies.dat
-x coords.cdf
&end
&speval
data Conf*.oout
&end
This may look distressingly simple: all you need is a topology. A report
on the process will be piped concat.out, a file equivalent to
sander's mdout, while energies go into the "dump"
file and coordinates get written to what coords.cdf, a
trajectory like you would get in sander's default
mdcrd. Inside the &speval module the only item
that appears is a request to find files of the name format
Conf*.oout. The trick here is to run this inside the directory
where you've made those conformations; otherwise just extend that name format
with the relative or absolute path and you're good to go. If you have
multiple directories full of files, add more data entries. This
operation elegantly reduces the hundreds of files created by conformational
search and ORCA evaluations to just two files:
coords.cdf, and energies.dat. Repeat for MPD.
This tarball contains the
distilled results for glycerol and MPD in separate directories.
To summarize Stage 1: We created a large number of conformations of
two molecules, glycerol and MPD, which share a lot of chemical features and
thus Amber (GAFF) atom types. We computed the MP2 single point energies of
each of these conformations and compiled the coordinates into a compact, NetCDF
format trajectory (for precision).
In practice: There are a lot of ways to improvise here, so long as
you roll out of Stage 1 with a list of structures and the energies they
correspond to according to your benchmark method (here, MP2 / cc-pvDZ). The
next stage, and the mdgx ¶m module, are agnostic to the
origins of this information. In this tutorial I have tried to impart what I
feel are "best practices" by doing all of this in the context of the gas phase
charge set. However, if you don't want to fit your own charges and have some
that are descriptive of the solution phase instead, it's not a terrible thing
to just roll with those.
For completeness: It is important to understand that the
conformations mdgx has just produced are optimized with respect to
the guess force field at this stage. The partial charges will not be
changing due to anything that happens in this tutorial, and in practice it is
sensible to make charges and then take them as given in order to make bonded
parameters. But, in this example, we are performing MP2 single point
calculations on GAFF-optimized structures. This business of getting the QM
energies of MM-optimized structures doesn't sit well with some people, and I
have every sympathy: it's not ideal in my mind, either. However, as I showed
in the 2014 paper
it is probably the lesser of two evils. Those who disagree with my approach
prefer to take MM-optimized structures and make their relative MM energies
agree with the QM single point energies of similar QM optimized structures:
these QM-optimized structures will typically have identical coordinates in the
handful of parameters that are being fitted, but the other degrees of freedom
have been optimized with respect to the QM and MM force fields, respectively.
If one wishes to pursue the other route, it is still possible with
mdgx: all you need is a set of coordinates and energies, the code
doesn't care how you got them.
Go on to Part 2
|


