


The AMBER ff15ipq-m Force Field Tutorial
Learning Outcomes
Introduction
This tutorial is designed to accompany the AMBER ff15ipq-m force field for protein mimetics [Bogetti et al. 2020]. This tutorial will guide you through the process of building a short tetrapeptide using tleap, resources for modifying an existing protein, and visualization of the simulation results. AMBER ff15ipq-m files required for this tutorial are included in AmberTools 21, as well as in AmberTools 20 Update.10 and Update.14.
Background
The AMBER ff15ipq-m force field is an expansion of the ff15ipq force field [Debiec et al. 2016] for the treatment of protein mimetics, including the D-α-, Cα-methylated α- or aminoisobutyric acid (Aib), β3, and cyclic β- (βcyc) residues, aminopyrrolidine carboxylic acid (APC) and trans-2-aminocyclopentane-1-carboxylic acid (ACPC). This force field is the latest in the lineage of Implicitly Polarized Charge (ipq) protein force fields with atomic charges that are implicitly polarized, meaning that the charges are intended to reproduce the mean field electron density of a molecule in explicit solvent. Consistent with the ff15ipq force field, the explicit SPC/Eb (extended simple point charge) water model was chosen for ff15ipq-m to take advantage of the model’s reproduction of experimental rotational protein diffusion in solution and the relatively low cost of a three-point water model compared to other four-point alternatives. The ff15ipq-m/ff15ipq force fields exhibit the following features: (i) reasonable propensities for salt-bridge formation (addressing the overstabilization of salt bridges by many force fields [Debiec et al. 2014]); (ii) a complete rederivation of all parameters to yield the expected balance of secondary structures for both globular proteins and non-globular (“disordered”) peptides; and (iii) the ability to calculate reasonably accurate NMR observables such as J-coupling constant for peptides and NMR relaxation for methyl groups.
Like other AMBER force fields, the use of ff15ipq-m in the AMBER software requires three major files: leaprc.mimetic.ff15ipq, frcmod.ff15ipq-m, and ff15ipq-m.lib. To enable the modeling of various unnatural amino acid residues, the following new residue and atom names have been created in the ff15ipq-m force field:
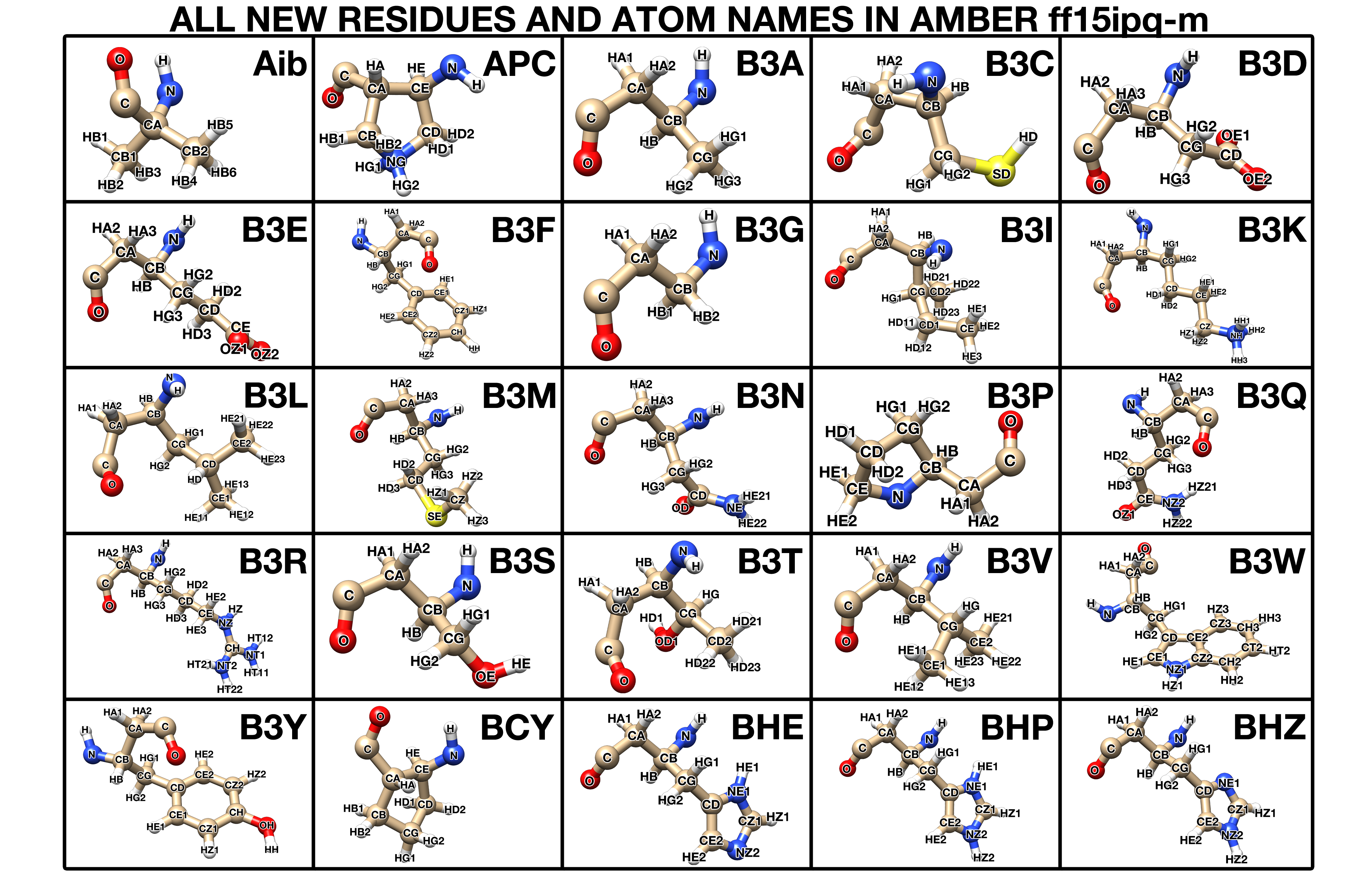
Figure 1.1: The new residue classes and atom names in AMBER ff15ipq-m. Click for larger version.
These new residue and atom names involve the following new atom types:
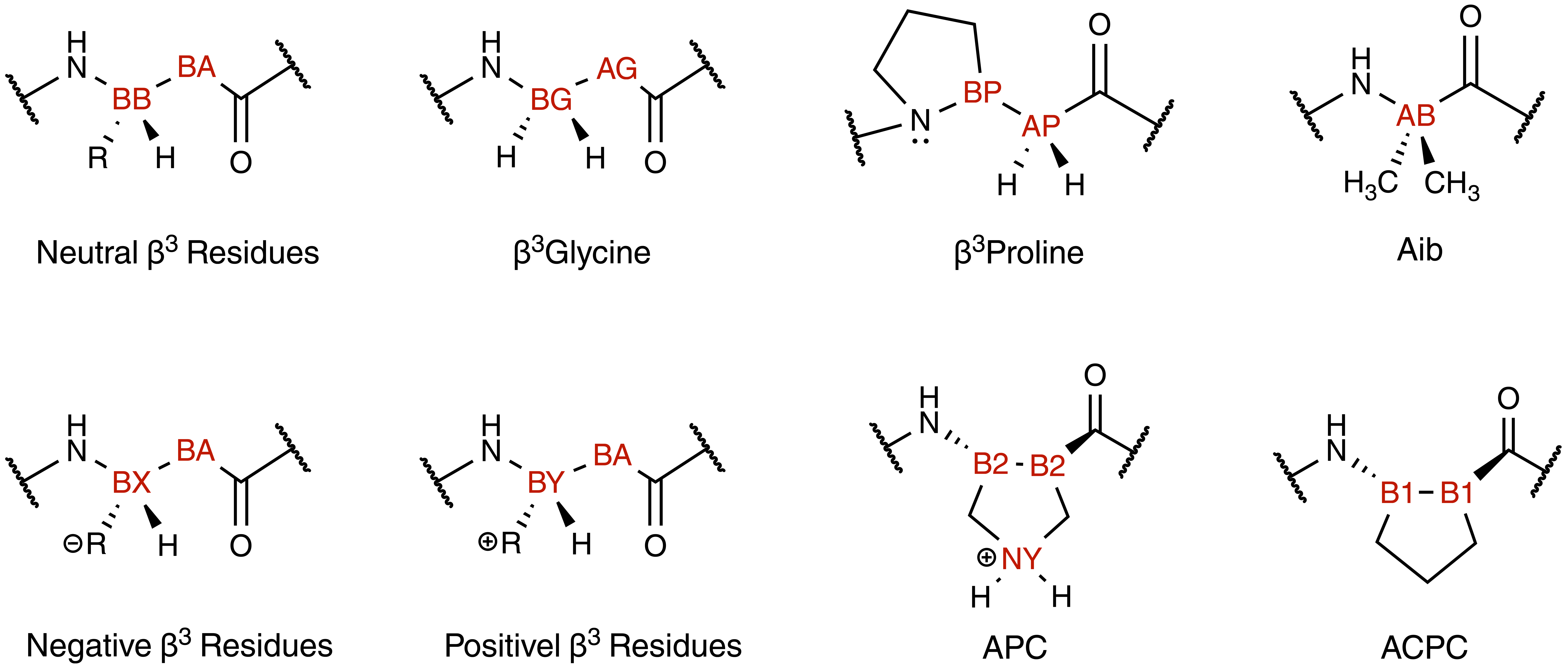
Figure 1.2: New atom types for artificial backbone units. The nomenclature for the new atom types (highlighted in red) is BA for β3-α-carbon, BB for β3-β-carbon, BG for β3Gly, etc.
Part 1: Building a Tetrapeptidomimetic
In this part of the tutorial, we will build a β3Alanine (B3A) tetrapeptide mimetic (peptidomimetic) and solvate using the water model (SPC/Eb) intended for the ff15ipq-m force field. First, let’s create a tleap input file (tleap.in):
source leaprc.mimetic.ff15ipq
source leaprc.water.spceb
mol = sequence { ACE ALA B3A ALA NME }
solvateOct mol SPCBOX 12.0
saveAmberParm mol B3A_solv.prmtop B3A_solv.inpcrd
savePdb mol B3A_solv.pdb
* Note: expect the loading times for the mimetic library files to take a few minutes.
Now run tleap with the input file:
$ tleap -f tleap.in
There will be three files that are created as a result of this command. B3A.prmtop is the parameter/topology file for the solvated β3Alanine system, B3A.inpcrd is the initial trajectory and coordinate information, and B3A.pdb is just the coordinates for visualization to make sure that our tetrapeptide checks out structurally.
If you do not already have a PDB file of the tetrapeptidomimetic, you can create one using the ambpdb program (see page 592 of the Amber 2020 Manual for details about ambpdb):
$ ambpdb -p B3A.prmtop -c B3A.inpcrd > B3A.pdb
We can visualize our B3A peptide pdb file with any molecular viewer such as Chimera or PyMol. VMD is useful for viewing either the pdb file alone, or the prmtop and inpcrd files together. At this stage the peptide should look like this (truncated 12Å octahedral water box is omitted for clarity):
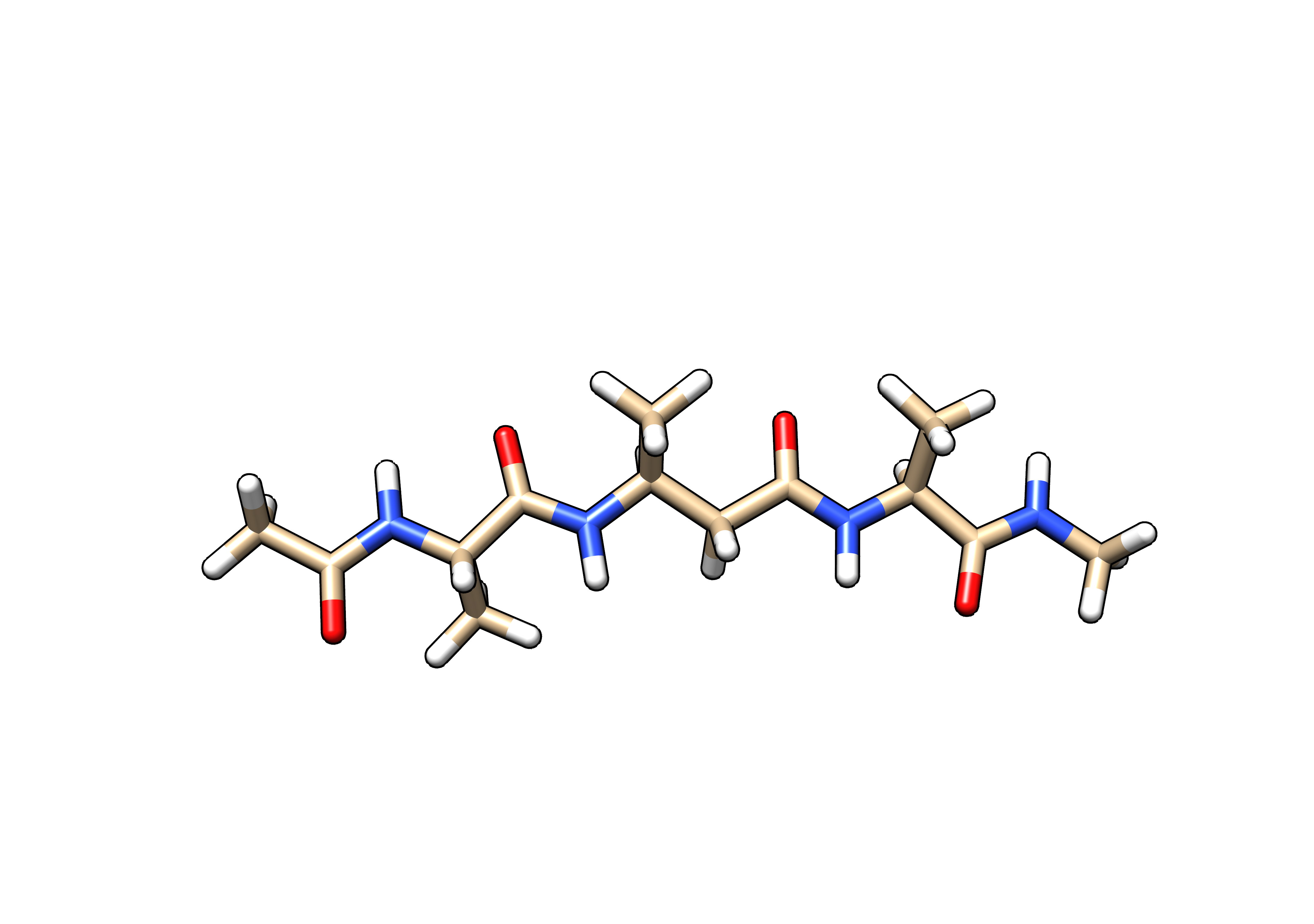
Figure 1.3: β3Alanine (B3A) tetrapeptide mimetic (peptidomimetic) solvated using the water model (SPC/Eb) intended for the ff15ipq-m force field.
Files from this stage can be accessed here: B3A_tleap.in B3A_solv.prmtop B3A_solv.inpcrd B3A_solv.pdb
Part 2: Modifying a Protein to Include an Unnatural Amino Acid
We will now prepare to simulate a protein with an unnatural amino-acid substitution, using the B1 Immunoglobulin-binding domain of streptococcal protein GB1 (PDB: 1PGB) as an example. We will substitute the Ala48 residue to the unnatural Aib residue by modifying the C-α hydrogen atom into a methyl group. To modify the Ala48 residue, we will use Chimera (Build Structure tool). Alternatively, you can use PyMol (Builder tool or a custom script) or VMD ( Molefacture plugin; see tutorial). For more information regarding building an AMBER compatible protein system in explicit solvent, see this Tutorial 1.4 Building Protein Systems in Explicit Solvent.
The ALA 48 to AIB 48 modification step in Chimera looks like this:
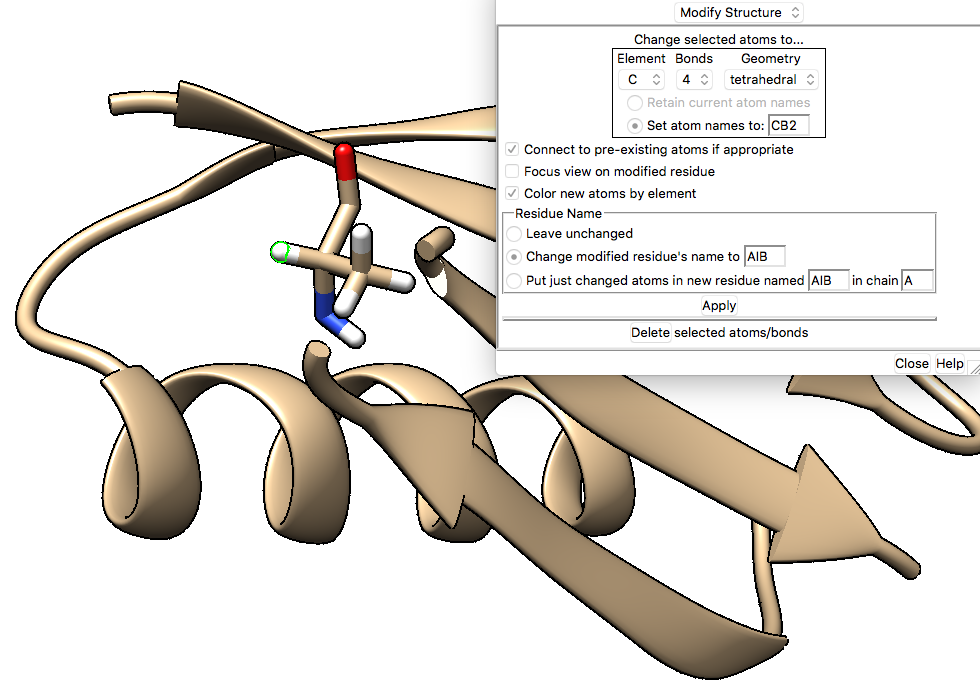
Figure 2: Changing 1PGB ALA 48 to AIB using the build-modify structure tool in Chimera.
We will save this new structure as 1PGB-m.pdb and double check the PDB file for correct residue and atom names. Make sure that the residue name for Ala48 is now AIB instead of ALA, the record type (column 1) is changed to standard (ATOM) instead of non-standard (HETATOM), and that the atom names are consistent with the ff15ipq-m library and frcmod file naming conventions. Visual representations of these can be found at the start of this document.
We can also prepare to simulate a protein from the PDB that already has artificial amino acids in its sequence such as 5HI1(PDB: 5HI1), which is a GB1 variant with Aib24, β3Lys28, β3Lys31, and Aib35. For this protein mimetic, follow the same protocol as before, adjusting the atom coordinates to standard ATOM types, changing the atom names in the PDB file to match the above mimetic residue guidelines, and following standard protocols to prepare your system conditions: Tutorial 1.4.
Now that we have AMBER-ready input files for two protein mimetic, let’s proceed with the solvation step in tleap:
source leaprc.mimetic.ff15ipq source leaprc.water.spceb GB1 = loadPdb 1PGB-m.pdb solvateOct GB1 SPCBOX 12.0 addIons Cl- 0 addIons Na+ 0 saveAmberParm GB1 GB1_solv.prmtop GB1_solv.inpcrd savePdb GB1 GB1_solv.pdbFinally, check the newly solvated structure in VMD or Chimera before moving forward with the simulation steps. Files from this stage can be found here: 1PGB.pdb 1PGB-m.pdb 5HI1.pdb 5HI1_solv.prmtop 5HI1_solv.inpcrd 5HI1_solv.pdb GB1_solv.prmtop GB1_solv.inpcrd GB1_solv.pdb
Part 3: Simulating the unnatural amino acid incorporated system
Now that we have prepared modified peptide and protein systems using the ff15ipq-m library and force field files, they can be simulated just like any other peptide or protein with the sander or pmemd modules on AMBER. For instance, we could utilize our files and follow the AMBER alanine dipeptide tutorial.
If we were interested in a more thorough analysis of our peptidomimetic system, it would be advisable to look at a free energy profile along the φ/ψ angles. This can be done by adapting the AMBER umbrella sampling tutorial to our peptidomimetic system. Since β-residues such as B3A have an extra backbone carbon, there is another angle to consider as well, referred to as the θ angle.
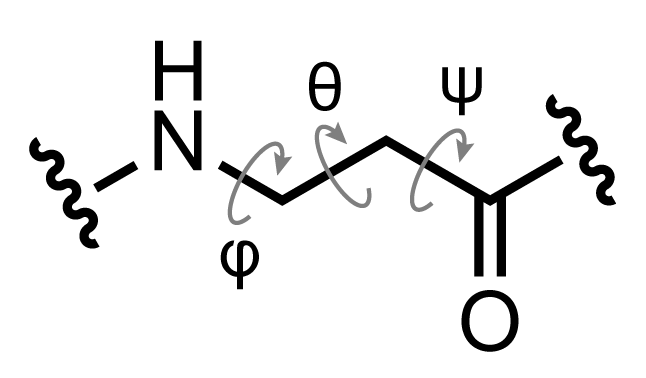
Figure 3: Definition of backbone torsion angles φ, θ, and ψ for the β-residue classes.
Ramachandran plots of all 25 ff15ipq-m residue classes using φ, θ, and ψ angles for umbrella sampling and subsequent free energy profile recovery with WHAM are available in the accompanying ff15ipq-m publication. Note that for visualizing non-standard residues, VMD does not register ribbons, cartoon, tube, trace representations because of limitations of the STRIDE program used to calculate secondary structure. It is recommended to instead use Chimera for those representations.
By Jeremy Leung and Darian Yang