


Building and Equilibrating a Membrane System with PACKMOL-Memgen
by Stephan Schott-Verdugo (s.schott-verdugo@fz-juelich.de)
Introduction
This tutorial will show you alternative ways of building a membrane system to perform molecular dynamics simulations within Amber, with or without an embedded protein, and with the optional inclusion of ligands. This is not a trivial task, as membrane environments are anisotropic (i.e. they are not the same in every dimension of the system), and, compared to typical explicit water simulations, the initial condition is important to obtain a representative structure in a reasonable simulation time.
Overview
In this tutorial we will assume that you are acquainted with the subprograms from Amber to perform conventional MD simulations, as well as with antechamber to derive parameters for simple molecules. We encourage you to check the previous membrane simulations tutorial; in this tutorial we will use Lipid21 with ff14SB, and even when in the previous tutorial Lipid14 was used, the philosophy of the force-field is the same. Lipid21, which includes corrections for torsion terms and adds support for sphingomyelins has been recently published and included in Amber 22; the same guidelines apply to all of the lipid force field included within Amber.
Prerequisites
To be able to use the latest functionalities, you should have Amber together with an updated AmberTools 23 or newer. To visualize the trajectories, we recommend to have VMD 1.9.3 and PyMOL for checking pdb structures.
Force field description
Currently, Amber includes Lipid21[9] as the main membrane force field, which, like Lipid14 [1] and Lipid11 [2] is based on modular parameters for phospholipid headgroups and acyl tails. In practice, this means that every headgroup and tail is an independent "residue", with each traditional phospholipid formed by an "N-terminal" tail, a central headgroup and a "C-terminal" tail [Note: Of course there is no N- or C-terminus in a lipid! This is just for you to understand that we are taking the same constructs as in a protein]. This has the advantage of leaving room to mix any headgroup with any pair of tails the user desires, while also allowing to develop new parameters for each part independently, without having to consider every combination a priori. To show this graphically, we will use DOPC (dioleoylphosphatidylcholine, or formally 1,2-dioleoyl-sn-3-glycero-phosphocholine) as an example:
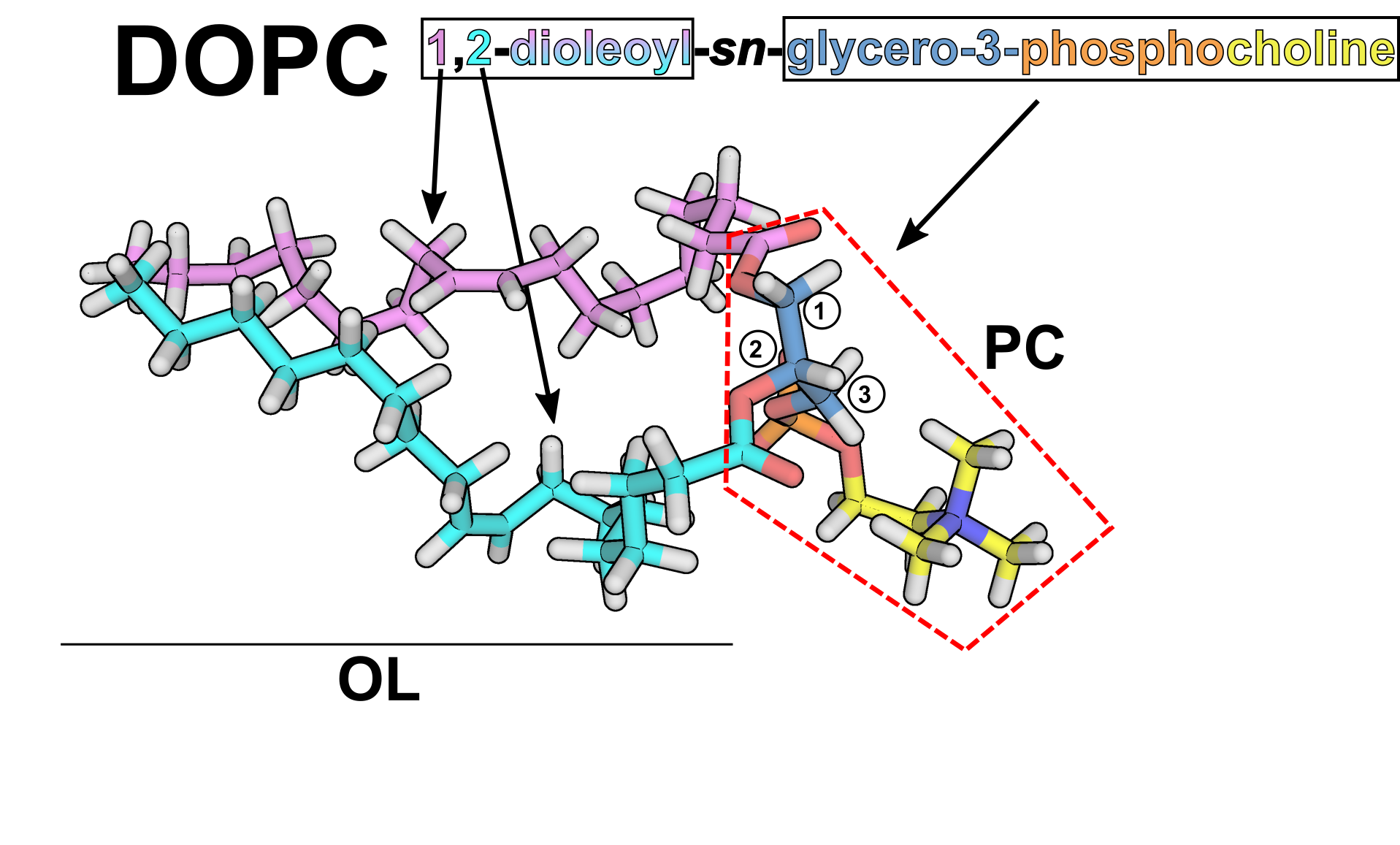
You can see the two aliphatic tails in pink and cyan,
corresponding to two "OL" residues, and the headgroup enclosed
in red dashed lines, which corresponds to a "PC" residue. Note
that the headgroup residue includes the ester groups connecting
the aliphatic tails. The connection between the tails and the
headgroup is treated analogously by LEaP as in the connection
between residues in a protein.
WARNING!: In practice,
this means that in the PDB you use with LEaP, there should be
a TER flag in-between lipids, but not in-between residues!
You have to be careful with this, as for example LEaP when
using Lipid17 added TER flags in-between residues of saved
PDBs if they are not part of a protein.
The Amber lipid force fields follow the ff99 parametrization
strategy, making it compatible with most Amber force fields. The
list of natively supported residues in Lipid21 can be found here.
We have extended this list of residues (in what we call
Lipid17ext) within PACKMOL-Memgen to include lysophospholipids
and phosphatidylinositols with different protonation states as experimental parameters (this
means, provided with no warranty at the moment). You
can find a list to all these residues in the Amber Manual, Section 12.6.
A note on lipid composition
Ok, now you should have an idea how the lipids are represented into the Amber force field, and what lipids you can include in a simulation. But probably you would like to simulate a particular protein of a defined organism, or study the membrane properties of an organelle. What membrane composition should you consider? This is, unexpectedly (for the newcomer at least), a very tricky question, and as usual, it depends in what you want to study. First, I would recommend to get some insights into the in vivo conditions of the system of interest.
A good starting point is the review from Harayama and Riezmann [3], where you should get a feeling of the different membrane components in general. An amazing web is the compendium assembled by Dr. William Christie from the James Hutton Institute/Mylnefield Lipid Analysis/LIPID MAPS [4]. This can be a great starting point to get all kind of information regarding lipids, from nomenclature to function and composition of different organism. A quick look will reveal that membranes are quite complicated, with different ratios of headgroups for different organisms or organelles, and different tails for each of the present headgroups depending also on the organism, but also on environmental factors, like temperature.
If you have seen publications where membrane environments are studied, you might recall that usually lipids like DOPC or POPC are used as the only component of the bilayer, both in simulations and in the lab. There is a reason for this: many proteins behave similarly regardless of the membrane environment, or at least it is considered a fair approximation. The more lipid types you include, things get more complicated (both in the lab, and in the simulation world), with the compromise of going farther away from the "real" system. Particularly for molecular dynamics simulations, you have to consider that the time required to relax a membrane system is (at least) in the order of hundreds of nanoseconds, which is reasonable for nowaday standards, but is still a considerable amount. Of course, this is not applicable for every system, and many proteins depend on the presence of particular lipids, like negatively charged headgroups or determined phosphatidylinositols.
Which composition should you use then? Hopefully the
minimal composition that has the most important components for
your system (and let's cross our fingers that you have
some information regarding this). This will hopefully allow it
to settle in a reasonable amount of time. You can always just
try with DOPC/POPC to see how things develop.
How to pack a membrane system?
There are multiple tools that you can use to set up a membrane
simulation system. One of the most used is CHARMM-GUI, and you
can find instructions on how to use it in
this tutorial and here.
Since AmberTools 18 though, we include PACKMOL-Memgen, which is
a workflow that automates the generation of membrane systems
using PACKMOL as the packing engine. Some advantages of
PACKMOL-Memgen are that you can script its usage for multiple
systems, run multiple instances in parallel without having to
worry about your browser getting stuck, add ligands to the
different phases and generate complex membrane compositions in a
single line. I will walk you through 3 systems which should be
representative of quite common usage scenarios:
-
1) a membrane-only system,
2) a protein embeded in a bilayer, and
3) the free diffusion of a ligand in a membrane environment.
Generating a membrane-only system
The thing with membrane systems, is that membranes by themselves are already interesting and complicated enough. You have virtually infinite possible combinations of lipids, and each will behave differently depending on the composition you choose, forming microdomains, affecting the width or curvature of the membrane, just to name a few. Of course a more limited subset is representative of the membranes you are able to find in vivo, but many would like to simulate things that are not necesarilly findable in real organisms. Let's say that you are interested in studying the inner-membrane of Gram-negative bacteria. This will change (again) from bacteria to bacteria, but a simplified representation of such a system is considering a composition of PE:PG 3:1 (at least for E. coli [6]). You will find this information regarding the headgroup ratios but not about the concrete phospholipids multiple times in the literature, leaving room to interpretation regarding the aliphatic tails that are bound to that particular headgroup composition. In this case, we will use DOPE:DOPG 3:1. To do this, you can use the following command:
packmol-memgen
--lipids DOPE:DOPG --ratio 3:1 --distxy_fix 100
--parametrize
where,
| --lipids DOPE:DOPG | specifies the lipids to use |
| --ratio 3:1 | sets the ratio of DOPE with respect to DOPG. The order depends on the order used in --lipids |
| --distxy_fix 100 | sets the dimensions in the membrane (xy-)plane to 100 Å |
| --parametrize | automatic parametrization. Uses Lipid21 and FF14SB by default. |
If you want to try with a different lipid composition, check the available lipids by calling packmol-memgen --available_lipids (or --available_lipids_all for an exhaustive list). Running the previous command will generate bilayer_only.pdb , which is the output of Packmol with AMBER residue names. Below you can see a rendered representation of the packed system. You might notice the high quantity of ions included, despite not using the --salt flag. This is due to the counterions added to compensate for the negative charges introduced by the DOPG headgroup, which in the volume used would represent a concentration higher than 0.15M!
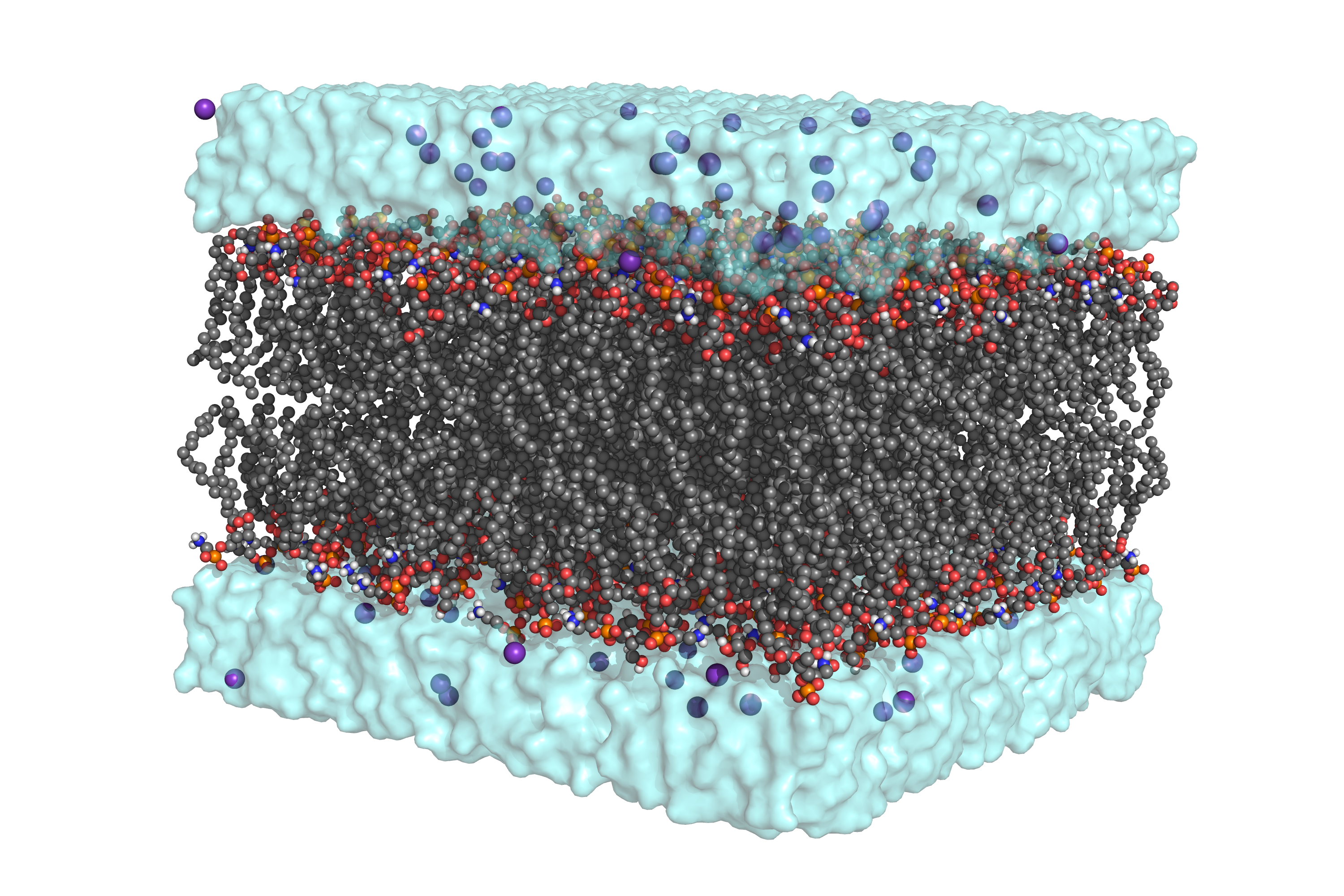
Since we requested the system to be parametrized, we will also
get:
bilayer_only_lipid.pdb
PDB file made by LEaP
bilayer_only_lipid.top
Topology file.
bilayer_only_lipid.crd
Coordinate input file.
You can directly use these files with the equilibration and minimization files described here.
Packing a protein into a membrane
In practice, packing a protein into the membrane is not much more complicated than what we already described. In this case, we will be packing the potassium channel KcsA (PDB: 1BL8). If you want to include a protein into a membrane system, the first thing that you have to consider is how it is oriented with respect to the membrane slab. By default, PACKMOL.Memgen will assume that your protein is not oriented, and will use memembed to place it properly. Despite it being a very handy tool for average proteins, if your membrane protein is very big or represents a complicated system, I recommend you to use the PPM server from OPM. If you use this webserver, you have to let the workflow know about this with the --preoriented flag!
Whatever protein you will be packing, you have to take care to have the proper protonation states, residue names, and caps in the PDB file. You can make the proper changes before performing the packing, taking care that the workflow will not change the protonation state you want (check --notprotonate and --nottrim), or make them before passing the packed file to LEaP. There are some tools that can help you in this process; you can obtain pdb2pqr in a local conda environment and use it with packmol-memgen with the --pdb2pqr flag, or use the PDB2PQR server (making sure to select AMBER as the naming scheme), MOE or Maestro, but you have to be aware that the hydrogens and cap names added match the names used in the force field. This case is a good example, as it requires some awareness from the user side to prepare the simulations; E71 of the KcsA channel has been shown to be in a protonated state at pH 7 [7], so we will change the residue name in the PDB file to match the proper protonation. We start with:
wget https://include.rcsb.org/download/1BL8.pdb
This will download 1BL8 from the PDB. Open the file with your preferred text editor and molecular viewer, and notice that there are 4 chains (from A to D), that potassium ions and a water molecule co-crystallized in the channel, and that each chain has the aforementioned GLU 71, whose side chain atoms are in fact missing. You can fix these with the softwares mentioned before, or let LEaP add them and let the MD sample a proper position. The latter might work as long as the amount of atoms to add is only discrete. We can change the residue for its protonated version by changing the residue name to GLH, the AMBER residue name for a protonated GLU, before packing the protein. The issue with this is that in PACKMOL-Memgen, proteins are by default deprotonated and protonated again with reduce, which currently does not recognize residue names of alternate protonation states, and will add wrongly named hydrogens. This will work as long as you remove the wrongly added hydrogens after the packing and before using LEaP (in any case, you will get an error message from LEaP pointing you to the wrong hydrogens if you try). An alternative is to run the packing without changing the residues, and change them after the packing:
packmol-memgen --pdb 1BL8.pdb --lipids DOPE:DOPG --ratio 3:1 --keepligswhere,
| --pdb | flag to specify the protein to include in the packing |
| --lipids DOPE:DOPG | specifies the lipids to use |
| --ratio 3:1 |
sets the ratio of DOPE with respect to DOPG. The order depends on the order used in --lipids |
| --keepligs | will keep non-proteic molecules (potassium and water in this case) in the PDB, usually removed by memembed. |
After the previous command finishes successfully, you will have the packed pdb bilayer_1BL8.pdb. You can inspect it with you preferred molecular viewer and check how the generated membrane looks like, and if any lipid is overlapping heavily with the protein, or if any of them got trapped in the protein channel. If you open the file with a text editor, you will notice that the residue numbers were changed; residues 71 in each chain, are now numbered 49, 146, 243 and 340. We can rename them to GLH with the following command:
for i in " 49" 146 243 340; do sed -i "/GLU . $i/s/GLU/GLH/g" bilayer_1BL8.pdb; done
WARNING! Please note that this step is completely system specific. It could be that your protein does not require any special treatment, or that you have all hydrogens properly added beforehand, in which case you can directly parametrize it. In this case I am assuming that I start from a not-ready-to-go protein from the PDB for instruction purposes! The way you prepare your own protein will depend completely on you.
Please check the file after executing the commands and see that the residues were properly renamed. It should look something like this.

Now, we can use LEaP to generate the topology and MD input coordinates. You can do this with your own script, or use the suggestion generated by PACKMOL-Memgen:
packmol-memgen --pdb 1BL8.pdb --lipids DOPE:DOPG \
--ratio 3:1 --keepligs --parametrize
You might have noticed that this is the same command as before, but with the included --parametrize flag. The script will identify that bilayer_1BL8.pdb is already in the folder, and will skip the packing process, and go straight to the parametrization step. If you want to be able to modify the LEaP script used, you can also run the script with the --keep flag, which will leave all the intermediate include including the leap.in file. If everything went well, you will end up with the following additional include:
bilayer_1BL8_lipid.pdb PDB file made by LEaPbilayer_1BL8_lipid.top Topology file.
bilayer_1BL8_lipid.crd Coordinate input file.
You can now run the minimization and
equilibration protocol using these files.
Adding free diffusing ligands to a (membrane) system
One potential application of MD simulations is to find binding sites or equilibration distributions for particular ligands of interest. PACKMOL-Memgen has some functions to help you in building a system to perform such simulations. In this case, we will use the same KcsA channel as an example. Let's say that for some reason you are interested in checking out the described blockage of the channel by tetraethylammonium (TEA) [8]. The first thing would be to obtain proper parameters to describe TEA; in this case we will use ESP charges from a single optimized conformation with Gaussian 09 with the RESP functions from Antechamber, and the GAFF2 forcefield. Here you can find the Gaussian output used, and the leap script to make the library file (check the antechamber description in the manual and in the tutorial here):
antechamber -i TEA.gout -fi gout -o TEA_resp.mol2 -fo mol2 \
-c resp -rn TEA -pf y -at gaff2
parmchk2 -i TEA_resp.mol2 -f mol2 -s 2 -o TEA.frcmod
tleap -f leap_TEA.in
As a result of the previous steps we will have TEA.pdb, TEA.lib with the parametrized sample topology and charges, and TEA.frcmod with parameters missing from GAFF2 that were estimated for completeness. Note that the frcmod is, for this particular case, empty; we still need it for running the script properly. With all these include, you can already perform the packing just as in the previous section, but adding theflags to include TEA into the system. An example would be:
packmol-memgen --pdb 1BL8.pdb --lipids DOPE:DOPG --ratio 3:1 --keepligs \
--solute TEA.pdb --solute_con 4 --solute_prot_dist 10
where,| --solute | specifies the PDB to be added as a solute |
| --solute_con | specifies the concentration to be added (to the water volume by default; check --solute_inmem). Number of molecules, or concentration if the M or % suffixes are added (e.g. 0.1 M). |
| --solute_prot_dist | sets a cylindrical constraint around the protein where the ligand can not be placed. Avoids starting with a bound configuration. |
This will generate a file called bilayer_1BL8.pdb that will include the TEA molecule in the water phase; open it with your preferred molecular viewer to inspect it. Particularly when adding only a handful of molecules, it could be that they end up in tiny cluster on either side of the membrane, which is due to the way Packmol handles the initial guess. If this is of concern for your molecules (maybe they are hydrophobic on the first place), a trick is to pass the --solute --solute_con flags multiple times (i.e. for this case, --solute TEA.pdb --solute_con 2 two times).As before, we did not request the script to parametrize the system straight away, to fix the GLU residues before passing the PDB to LEaP; the same sed commands will do the trick:
for i in " 49" 146 243 340; do sed -i "/GLU . $i/s/GLU/GLH/g" bilayer_1BL8.pdb; done
After which, we need to obtain the topology. For this, PACKMOL-Memgen can also generate the LEaP input:
packmol-memgen --pdb 1BL8.pdb --lipids DOPE:DOPG --ratio 3:1 --keepligs \
--solute TEA.pdb --solute_con 4 --solute_prot_dist 10 \
--ligand_param TEA.frcmod:TEA.lib --gaff2 --parametrize
which is the same as before, but including the
--parametrize
flag and:
| --ligand_param | specifies the frcmod and lib file of ligand included in the system (either in the original PDB or added as solute). It has the shape FRCMOD:LIB without spaces in between. |
| --gaff2 | sets GAFF2 as the forcefield to be included during the LEaP run. |
After the parametrization finished, you will end up with the
following include:
bilayer_1BL8_lipid.pdb
PDB file made by LEaP
bilayer_1BL8_lipid.top
Topology file.
bilayer_1BL8_lipid.crd
Coordinate input file.
You can use these files with the minimization and equilibration
scripts described here.
Additional tools
There are additional tools included within PACKMOL-Memgen that are not described here, like being able to parametrize protein/membrane systems with the coarse-grained SIRAH force field (check the flag --sirah), adding mixed solvents in addition to water (check --available_solvents), making curved membranes (check --curvature and --xygauss) or solvating systems without a membrane (check --solvate & --cubic). You are encouraged to experiment and try the available flags (check --help)!!!
References
[1] Dickson, C.J., Madej, B.D., Skjevik, A.A., Betz, R.M., Teigen, K., Gould, I.R., Walker, R.C., "Lipid14: The Amber Lipid Force Field", J. Chem. Theory Comput., 2014, 10(2), 865-879.DOI: 10.1021/ct4010307
[2] Skjevik, A.A,; Madej. B.D.; Walker, R.C.; Teigen, K.; "LIPID11: A Modular Framework for Lipid Simulations Using Amber", Journal of Physical Chemistry B, 2012, 116 (36), pp 11124-11136.
DOI: 10.1021/jp3059992
[3] Harayama, T., and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nature Reviews Molecular Cell Biology, 19(5), 281-296.
DOI: 10.1038/nrm.2017.138
[4] Christie, W. W., The LipidWeb, https://lipidmaps.org/resources/lipidweb/lipidweb_html/index.html
[5] Schott-Verdugo, S., & Gohlke, H. (2019). PACKMOL-Memgen: A simple-to-use generalized workflow for membrane-protein/lipid-bilayer system building. Journal of chemical information and modeling. DOI: 10.1021/acs.jcim.9b00269
[6] Murzyn, K., Róg, T., & Pasenkiewicz-Gierula, M. (2005). Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophysical Journal, 88(2), 1091–1103.
DOI: 10.1529/biophysj.104.048835
[7] Bhate, M. P., & McDermott, A. E. (2012). Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proceedings of the National Academy of Sciences of the United States of America, 109(38), 15265–15270.
DOI: 10.1073/pnas.1211900109
[8] Guidoni, L., & Carloni, P. (2002). Tetraethylammonium binding to the outer mouth of the KcsA potassium channel: Implications for ion permeation. Journal of Receptors and Signal Transduction, 22(1–4), 315–331.
DOI: 10.1081/RRS-120014604
[9] Dickson, Callum J., Ross C. Walker, and Ian R. Gould. "Lipid21: Complex Lipid Membrane Simulations with AMBER." Journal of chemical theory and computation (2022).
DOI: 10.1021/acs.jctc.1c01217