


An Introduction to CPPTRAJ
By Daniel R. Roe, July 2014
TABLE OF CONTENTS
IntroductionLoading a Topology and Trajectory
Specifying an Action
Processing the Trajectory
Working With Data Sets
Running in Batch Mode
Associated Files
Introduction
This tutorial will give a brief overview of analyzing simulation data with CPPTRAJ. CPPTRAJ is the successor to PTRAJ, with many additional features. Some basic and common types of analysis will be covered, as well as the basics of data set handling in CPPTRAJ. This assumes that AmberTools has been successfully installed and has been tested. This also assumes some familiarity with Amber atom mask selection syntax. For more details on atom mask selection syntax see the Amber manual (section 28.2.3. Atom Mask Selection Syntax in the Amber 14 manual). In addition, xmgrace will be required to view some of the output data.
Throughout this tutorial a short example trajectory of the beta-hairpin trpzip2 will be used. The trajectory is in NetCDF format, which is faster to process, more compact, higher precision, and more robust than the ASCII format. NetCDF is enabled by default in Amber, but if you find that your CPPTRAJ cannot read this trajectory please contact the Amber mailing list for help. The trajectory and associated topology can be downloaded here:
- trpzip2.gb.nc ¦ MD5 sum: 059435cfe1fcd57c327d46711da9ceff
- trpzip2.ff10.mbondi.parm7 ¦ MD5 sum: 140044a45c683612da3431dfd0697e22
For more detailed information on CPPTRAJ, see here:
Loading a Topology and Trajectory
To start CPPTRAJ, type 'cpptraj' from the command line:
[user@computer ~]$ cpptraj
CPPTRAJ: Trajectory Analysis. V14.05
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
>
Running CPPTRAJ with no arguments brings up the interactive command line.
The command line is useful for running simple or short analyses. The
command line allows tab completion of file names and commands. Also, in
interactive mode all commands used are written to the file 'cpptraj.log'
(this name can be changed with the '--log
Before reading in a trajectory, CPPTRAJ needs to know what the system looks like. This information is contained with topology files. The first step is to load the topology file with the 'parm' command:
> parm trpzip2.ff10.mbondi.parm7 Reading 'trpzip2.ff10.mbondi.parm7' as Amber Topology
The topology has now been loaded. You can see what topologies are loaded with the 'list' command:
> list parm PARAMETER FILES: 0: 'trpzip2.ff10.mbondi.parm7', 220 atoms, 13 res, box: None, 1 mol
The output shows the topology index (which starts from 0) followed by some brief information on the topology. More detailed information can be obtained using the 'parminfo <#>' command, where <#> is the index of the desired topology.
> parminfo 0 Topology trpzip2.ff10.mbondi.parm7 contains 220 atoms. 13 residues. 1 molecules. 227 bonds (104 to H, 123 other). 402 angles (233 with H, 169 other). 853 dihedrals (481 with H, 372 other). Box: None GB radii set: modified Bondi radii (mbondi)
Now that the topology file is loaded, we can tell CPPTRAJ which trajectory we are going to process:
> trajin trpzip2.gb.nc Reading 'trpzip2.gb.nc' as Amber NetCDF
Note that this does not immediately read the trajectory, rather it places the trajectory in the input trajectory list for processing later. To see what trajectories are currently in the input trajectory list we can again use the 'list' command:
> list trajin INPUT TRAJECTORIES: 0: 'trpzip2.gb.nc' is a NetCDF AMBER trajectory, Parm trpzip2.ff10.mbondi.parm7 (reading 1201 of 1201) Coordinate processing will occur on 1201 frames.
Specifying an Action
Now that a topology and trajectory have been loaded, we can specify actions to generate data from the trajectory. Say for example we would like to know the end-to-end distance for the hairpin over the course of the trajectory. We can use the 'distance' command to get this information. First, we can use the 'help' command to remind us of the syntax for 'distance':
> help distance [<name>] <mask1> <mask2> [out <filename>] [geom] [noimage] [type noe] Options for 'type noe': [bound <lower> bound <upper>] [rexp <expected>] [noe_strong] [noe_medium] [noe_weak] Calculate distance between atoms in <mask1> and <mask2>
The 'help' command can be used with no arguments to bring up a list of all commands.
In order to figure out which atoms correspond with the end residues of trpzip2, we can use the 'resinfo' command:
> resinfo
#Res Name First Last Natom #Orig
1 SER 1 13 13 1
2 TRP 14 37 24 2
3 THR 38 51 14 3
4 TRP 52 75 24 4
5 GLU 76 90 15 5
6 ASN 91 104 14 6
7 GLY 105 111 7 7
8 LYS 112 133 22 8
9 TRP 134 157 24 9
10 THR 158 171 14 10
11 TRP 172 195 24 11
12 LYS 196 217 22 12
13 NHE 218 220 3 13
From this output we can see that our end residues are 1 and 13. In general, the 'resinfo', 'atominfo', and 'molinfo' commands are useful for examining your system layout and/or testing the result of an atom mask expression. For example, to see what atoms will be selected by the atom mask ':13' (residue 13):
> atominfo :13 #Atom Name #Res Name #Mol Type Charge Mass GBradius El 218 N 13 NHE 1 N -0.4630 14.0100 1.5500 N 219 HN1 13 NHE 1 H 0.2315 1.0080 1.3000 H 220 HN2 13 NHE 1 H 0.2315 1.0080 1.3000 H
We can now enter our 'distance' command:
> distance end-to-end :1 :13 out dist-end-to-end.agr
DISTANCE: :1 to :13, center of mass.
This says to calculate a distance named end-to-end from the center of mass of residue 1 to residue 13, writing the results to a file named 'dist-end-to-end.agr'. The file format will be xmgrace-readable because the filename extension '.agr' is recognized by CPPTRAJ as xmgrace. We could change the format to gnuplot-readable by specifying a '.gnu' extension instead. If the extension is '.dat' or not recognized, CPPTRAJ will default to a standard column format. For a complete list of supported formats and their associated extensions see the Amber 14 manual.
Note that similar to 'trajin', entering an action does not execute it right away. Instead, it has gone into the action list. To see what actions are currently present in the action list we can use the 'list' command:
> list actions ACTIONS: 0: [distance end-to-end :1 :13 out dist-end-to-end.agr]
Processing the Trajectory
We have now loaded a topology, a trajectory, and have specified an action. The command can now be executed by specifying 'run' or 'go'. This tells CPPTRAJ to process each loaded trajectory, executing any specified actions on each frame. During trajectory processing some information will be printed that describes what CPPTRAJ is doing. First, information on the currently loaded topologies and trajectories are printed:
> run ---------- RUN BEGIN ------------------------------------------------- PARAMETER FILES: 0: 'trpzip2.ff10.mbondi.parm7', 220 atoms, 13 res, box: None, 1 mol, 1201 frames INPUT TRAJECTORIES: 0: 'trpzip2.gb.nc' is a NetCDF AMBER trajectory, Parm trpzip2.ff10.mbondi.parm7 (reading 1201 of 1201) Coordinate processing will occur on 1201 frames. TIME: Run Initialization took 0.0000 seconds.
Note that if any reference coordinates or output trajectories are specified they will appear here as well.
Next, the first trajectory will be loaded and any actions will be set up for that trajectory/topology:
BEGIN TRAJECTORY PROCESSING: ..................................................... ACTION SETUP FOR PARM 'trpzip2.ff10.mbondi.parm7' (1 actions): 0: [distance end-to-end :1 :13 out dist-end-to-end.agr] :1 (13 atoms) to :13 (3 atoms), imaging off.
Here our 'distance' action has been set up.
Next, each frame in the trajectory will be read and processed:
----- trpzip2.gb.nc (1-1201, 1) ----- 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
When all trajectories have been processed, a summary of the total run will be written, including number of frames processed, any action-specific output, data sets generated, and data files written.
Read 1201 frames and processed 1201 frames. TIME: Trajectory processing: 0.0094 s TIME: Avg. throughput= 127372.9982 frames / second. ACTION OUTPUT: DATASETS: 1 data set: end-to-end "end-to-end" (double, distance), size is 1201 DATAFILES: dist-end-to-end.agr (Grace File): end-to-end ---------- RUN END ---------------------------------------------------
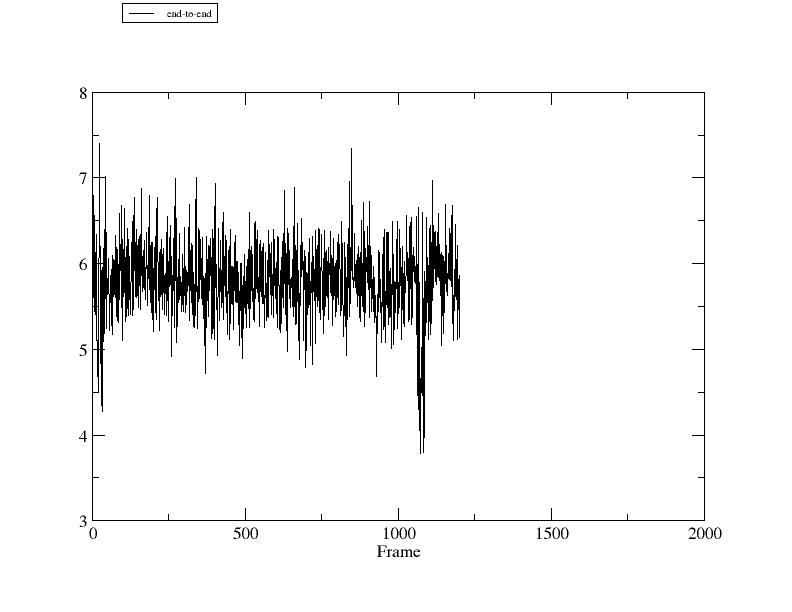
If xmgrace is installed and you are running and X-server, you can view the output right from the CPPTRAJ command line:
> xmgrace dist-end-to-end.agr
Working With Data Sets
From the 'DATASETS' section of the output:
DATASETS: 1 data set: end-to-end "end-to-end" (double, distance), size is 1201
we see that we have generated one data set named 'end-to-end', with the legend "end-to-end", that is a double-precision distance data set with 1201 elements. We can now continue to manipulate this data set if desired. Say for example you also want to write this data in the standard (column) data format. You can use the 'writedata' command like so:
> writedata end-to-end.dat end-to-end end-to-end
The linux 'head' command can be used directly from the CPPTRAJ command line to view the first few lines of 'end-to-end.dat'
> head end-to-end.dat
#Frame end-to-end
1 6.4251
2 5.9250
3 6.7926
4 6.3125
5 5.7580
6 5.4389
7 6.1086
8 6.5588
9 5.6949
Now that we are finished, type 'quit' or 'exit' to exit CPPTRAJ.
> quit TIME: Total execution time: 131.2112 seconds. -------------------------------------------------------------------------------- To cite CPPTRAJ use: Daniel R. Roe and Thomas E. Cheatham, III, "PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data". J. Chem. Theory Comput., 2013, 9 (7), pp 3084-3095.
Running in Batch Mode
Instead of running interactively, CPPTRAJ can also be run using one or more input files. Since the log file 'cpptraj.log' has recorded every command used, you can use the log file as the basis for a cpptraj "script". For example to generate everything we have done so far the following input could be used:
parm trpzip2.ff10.mbondi.parm7 trajin trpzip2.gb.nc distance end-to-end :1 :13 out dist-end-to-end.agr run writedata end-to-end.dat end-to-end
Paste the above commands into a file called 'cpptraj.in'. The commands can then be executed like so:
[user@computer ~]$ cpptraj -i cpptraj.in
CPPTRAJ: Trajectory Analysis. V14.05
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
INPUT: Reading Input from file cpptraj.in
[parm trpzip2.ff10.mbondi.parm7]
Reading 'trpzip2.ff10.mbondi.parm7' as Amber Topology
[trajin trpzip2.gb.nc]
Reading 'trpzip2.gb.nc' as Amber NetCDF
[distance end-to-end :1 :13 out dist-end-to-end.agr]
DISTANCE: :1 to :13, center of mass.
[run]
---------- RUN BEGIN -------------------------------------------------
The rest of the output will be similar to what was seen previously.
Note that it is VERY IMPORTANT to check your output for lines containing 'Warning:' or 'Error:' as these messages indicate there may be a problem with your input.
Associated Files
These files can be used to check your output.
dist-end-to-end.agrend-to-end.dat
cpptraj.in
Copyright Daniel R. Roe, 2014