Introduction
When preparing to write this note, I faced the following unavoidable choice: (1) use a simple and hence
relatively boring example that is easily runnable within an hour or (2) consider a larger system that
takes longer to run, yet is marginally more interesting. Option (2) won this time, and hence
this text describes two ways of simulating the Ace− (Pro)5 −NHe peptide in AMBER 11. All
the files and scripts needed to reproduce the runs described in this note should be available
alongside with this document in the
polyproline-tutorial-files.tar.bz2
archive (Warning: ≈ 60
Mb).
Preliminaries
We make the prmtop and inpcrd files using the teLeap program with the input shown in Fig.1. We are
going to use GB/SA solvation model from Refs.[2, 3, 4] (igb = 2 and gbsa = 1) and therefore line 5
instructs the teLeap program to use appropriate radii. The rest should be self-explanatory.
It is instructive to try regular molecular dynamics to see the lack of ergodicity at room temperature
(assumed to be 300K). To this end, one can use the mdin file shown in Fig.2. Apart from the usual &cntrl
namelist the file includes the ncsu_abmd section that defines a collective variable of type of
COS_OF_DIHEDRAL (lines 17–25) and instructs sander to save the values of the variable every 500
steps (1 ps) to the monitor.txt file (lines 12, 14, and 15). The collective variable is given
by the sum of cosines of the dihedral angles along the peptide bonds connecting successive
proline residues (see comments in Fig.2, lines 20–24). For all the bonds in cis conformation, the
variable would be assume the value of five, while for the all trans configuration, it would be
equal minus five. Once the simulation is complete, examination of the monitor.txt file shows
that no cis/trans transitions happened over one nanosecond. It is instructive to run it longer
and try higher temperatures as well. Summarizing, the regular molecular dynamics is not
ergodic in this case: the trajectory does not visit all equilibrium configurations with Boltzmann
probability; it gets stuck in the vicinity of the initial configuration because of the high (free) energy
barriers separating different (free) energy minima. The aim of this tutorial is to show two
possible routes to study the equilibrium ensemble in spite of presence of these barriers. In
the next section we are going to consider so-called steered molecular dynamics that can be
used to compute the free energy differences between different conformations. Afterwards we
are going to go through a more demanding (yet more powerful as well) replica-exchange[1]
technique.
1 **** this line is required ****
2 &cntrl
3 ntwx = 0, ntpr = 5000, ntwr = 50000,
4 ntt = 3, temp0 = 300.0, gamma_ln = 1.0,
5 igb = 2, gbsa = 1, dielc = 1.0, cut = 18.0,
6 ntb = 0, ntc = 2, ntf = 2, tol = 0.000001,
7 nstlim = 500000, dt = 0.002, ntp = 0, ibelly = 0,
8 ntr = 0, imin = 0, irest = 0, ntx = 1, ig = 27606
9 /
10
11 ncsu_abmd
12 mode = ANALYSIS
13
14 monitor_file = ’monitor.txt’
15 monitor_freq = 500 ! 1 ps
16
17 variable
18 type = COS_OF_DIHEDRAL
19
20 i = ( 2, 5, 7, 17, ! :1@CH3 == :1@C == :2@N == :2@CA
21 17, 19, 21, 31, ! :2@CA == :2@C == :3@N == :3@CA
22 31, 33, 35, 45, ! :3@CA == :3@C == :4@N == :4@CA
23 45, 47, 49, 59, ! :4@CA == :4@C == :5@N == :5@CA
24 59, 61, 63, 73) ! :5@CA == :5@C == :6@N == :6@CA
25 end variable
26 end ncsu_abmd
27
Figure 2: Input file for the sander program suitable for doing regular molecular dynamics at room
temperature.
polyproline-tutorial-files/2.preliminaries
directory.
Steered molecular dynamics
The so-called steered molecular dynamics amounts to a non-stationary harmonic restraint applied to a (set
of) collective variable(s). The method is extensively discussed in the literature and we therefore skip
any details here directing the reader to the excellent Refs.[5, 6, 7] and references therein.
A short summary for the lazy: one can “pull” the system between different states, measure
the non-equilibrium work performed, and then infer corresponding equilibrium free energy
differences.
Several subsystems in sander implement the “pulling” functionality and only one of them (ncsu_smd) is
exposed in this writeup. Curious reader is directed to the user manual to find out more on these and
related matters.
We proceed by pulling on the COS_OF_DIHEDRAL variable described above changing its value between -5 and
5 (that is, between the all-trans and all-cis conformations). To get a robust Δf we perform one hundred
of forward pullings (going from -5 to +5) and one more hundred of reverse pullings (going
from +5 to -5). Initial coordinates used for each of the ncsu_smd runs were equilibrated in the
corresponding state (all-trans for the forward pullings, and all-cis for the reverse pullings).
Relevant section of mdin file is shown in Fig.3 and should be self-explanatory. The progress of
the pulling is reported to the ’work.txt’ file every 10 ps. The total work is reported at the
end of this file once the simulation is complete. The files for all two hundred pulls can be
found in polyproline-tutorial-files/3.smd directory and we encourage the reader to examine
(or re-run) them. There is also a perl script that can be used to compute the free energy
difference.
Numerical values of the non-equilibrium work for the forward (WF ) and reverse (WR) pullings can be
used to compute the Δf estimate using Crooks[5] relations (the equation for the estimator coincides with
the earlier Bennet’s acceptance ratio formula[8]) from the following equation:
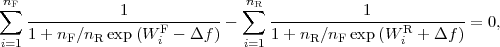 | (1) |
with nF,R denoting the numbers of forward and reverse simulations, respectively. We got 4.58 kcal/mol for
the Δf. It is instructive to compare this estimator with the ones computed from the Jarzynski[9]
identity applied to the only forward (10.65 kcal/mol) or only reverse (-1.49 kcal/mol) processes.
(Hamiltonian) replica exchange
The replica-exchange method is a powerful formulation first proposed by Geyer in 1991 in conference
proceedings[1] and subsequently rediscovered in a different area of science[10]. The basic idea of the
technique is to exchange configurations between several simulations in a way that preserves detailed
balance and hence makes every simulation to sample from its target distribution. The barriers that are
high in one replica (refered to as “cold” replica with respect to the barriers) may happen to
be significantly lower in another one (“hot” replica) so that the barriers crossing is greatly
enhanced.
The success of the method depends crucially on the choice of the “hot” replica(s). Here we are going to
use the potentials of mean force (PMF) to bias the dynamics in the “hot” replicas. The advantage of this
approach over the more conventional parallel tempering is that the temperature does not change across
the replicas hence rendering this kind of setup more suitable for explicit solvent simulations. We
(shamelessly) refer curious readers to our preprint http://arxiv.org/abs/0911.5132v2 for a more
comprehensive presentation.
The idea is very simple (and by no means ours): assuming that a “slow mode” can be described by a
collective variable σ(r), and that the potential of mean force (also referred to as Landau free energy)
associated with that collective variable is known, one can construct a “hot” replica by biasing the original
dynamics by the negative of the PMF thus rendering the states with different values of σ(r) equally
probable. In practice the PMF is typically unknown, yet fortunately, a biasing potential that is within a
few kBT of the true PMF, will do the trick. The latter can be computed in a variety of ways. In this
writeup we are going to use the so-called ABMD[11] (adaptively biased molecular dynamics), which
is yet another variation of the earlier local elevation[12] method (LEM), grown up from our
efforts to make the so-called metadynamics[13, 14] method (another LEM variation) more
practical.
For the Ace− (Pro)5 −NHe peptide, one readily identifies the cis/trans transitions of the peptide bonds as
the “slow modes”. There are five of them, and hence our aim is to setup a replica-exchange scheme with
five “hot” replicas biased by the PMFs associated with the corresponding dihedral angles, five more
replicas biased by the scaled down PMFs (to serve as proxies for more efficient random walk
between the replicas), and one not biased replica, from which we are going to “read” the
equilibrium samples. We compute the (approximate) PMFs using ABMD. For the very first, rough
approximation (initial condition for more demanding replica-exchange runs), we assume that the PMF
for the :1@CH3 == :1@C == :2@N == :2@CA dihedral of the Ace− (Pro)5 −NHe is close to the
same PMF computed for Ace−Pro−NHe, and that the PMFs for the dihedrals between Pro
residues in the Ace− (Pro)5 −NHe are close to the one computed for Ace− (Pro)2 −NHe
dimer.
We begin with the monomer and proceed by doing two-stage “flooding”: starting with more aggressive
(less equilibrium) and following it up with a smoother (closer to the equilibrium). Please find the
corresponding files in the 4.hremd/1.preliminaries/0.monomer subdirectory of the supporting files
archive. The “flooding” here means na�ve, linear grows of the biasing potential aimed to
compensate for the true PMF. We refer the reader to our preprint for the details. Finally,
we check the “quality” of the PMF by doing short equilibrium biased run (the files are in
0.monomer/3.umbrella). It turns out that the angle indeed explored all possible values in this short
run. This implies that the biasing potential is within few kBT of the true PMF, as intended.
If it turned out to be not the case, we would go with slower and slower flooding as long as
needed.
In the next step we compute PMF for the :2@CA == :2@C == :3@N == :3@CA dihedral of Ace− (Pro)2 −NHe
dimer following the same routine (the files in 4.hremd/1.preliminaries/0.dimer).
In order to equilibrate initial replica-exchange setup and to check whether those approximate PMFs
computed using smaller molecules do approximate the true ones with the accuracy not worse than a few
kBT, we ran a short replica-exchange simulation comprising ten replicas (five fully biased and five
“proxies”). These files are located in 4.hremd/2.hremd.eq and we encourage the reader to examine
them. We utilize the familiar multisander philosophy, yet use another (not mainstream) code
path which is activated by the ncsu_bbmd section in the mdin files. Everything there should be
self-explanatory (hit me by an e-mail, if it is not). This short run has revealed that the initial PMF
do not approximate the true ones very well, as the values of the dihedrals are distributed
very non-uniformly. To correct for that, we did a “flooding” run (4.hremd/3.hremd.flooding)
with stationary “proxy” biasing potentials. We then checked the (presumably) more accurate
PMFs by doing a short equilibrium run (4.hremd/4.hremd.umbrella) and proceeded to the
production phase (in case we where unhappy with the PMFs quality, we would do more of slow
“flooding”).
The “production” run is in 4.hremd/5.hremd.production. It comprises eleven replicas (five “hot”, five
“proxies” and one “cold”) each ran for 50 ns. We try to exchange five random pairs of replicas every 91
step. The exchange statistics is reported in the exchange_log_file and turned out to be quite
well.
Upon completion of the “production” run we got: (1) the monitor files from all eleven replicas: ten holding
biased samples of the dihedral angles, and eleventh with the equilibrium samples of the COS_OF_DIHEDRAL
collective variable; (2) unbiased configurations saved as the trajectory from the eleventh replica. We
are going to analyze the trajectory, noting in passing that the biased samples can be used
to improve the precision of the PMFs associated with the dihedral angles. We used ptraj
to extract the values of the slow angles from the trajectory. It can be seen that the angles
are indeed very sharply concentrated near the cis and trans values and hence they can be
unambiguously classified as being either cis or trans. It is thus natural to use five-letter string
consisting of T (for trans) and C (for cis) to describe each trajectory frame. A simple perl
script (omegas.pl file in the scripts subdirectory of the supplementary archive) can be used
to compare the frequencies of different conformations. We got that the TTTTT (all trans)
configuration is indeed the most probable one (global minimum) with the probability of 33%. It is
also possible to compute the free energy difference between the TTTTT and CCCCC states
(that would correspond to the COS_OF_DIHEDRAL values of -5 and +5 respectively): we got
4.42 kcal/mol, which compares very favorably with the steered molecular dynamics described
above.
References
[1] C. J. Geyer. Markov chain monte carlo maximum likelihood. In Computing Science
and Statistics: The 23rd symposium on the interface, pages 156–163, Fairfax, 1991. Interface
Foundation.
[2] A. Onufriev, D. Bashford, and D. A. Case. Modification of the generalized Born model
suitable for macromolecules. J. Phys. Chem. B, 104:3712–3720, 2000.
[3] A. Onufriev, D. Bashford, and D. A. Case. Exploring protein native states and large-scale
conformational changes with a modified generalized Born model. Proteins, 55:383–394, 2004.
[4] J. Weiser, P. S. Shenkin, and W. C. Still. Approximate Atomic Surfaces from Linear
Combinations of Pairwise Overlaps (LCPO). J. Comp. Chem., 20:217–230, 1999.
[5] Gavin E. Crooks. Path-ensemble averages in systems driven far from equilibrium. Phys.
Rev. E, 61(3):2361–2366, Mar 2000.
[6] Michael R. Shirts, Eric Bair, Giles Hooker, and Vijay S. Pande. Equilibrium free energies
from nonequilibrium measurements using maximum-likelihood methods. Phys. Rev. Lett.,
91(14):140601, 2003.
[7] Ioan Kosztin, Bogdan Barz, and Lorant Janosi. Calculating potentials of mean force and
diffusion coefficients from nonequilibrium processes without jarzynski’s equality. J. Chem.
Phys., 124(6):064106, 2006.
[8] C. H. Bennett. Efficient estimation of free energy differences from monte carlo data. J.
Comp. Phys., 22:245–268, 1976.
[9] C. Jarzynski. Nonequilibrium equality for free energy differences. Phys. Rev. Lett.,
78(14):2690–2693, 1997.
[10] K Hukushima and K Nemoto. Exchange monte carlo method and application to spin glass
simulations. Journal of the Physical Society of Japan, 65:1604–1608, 1996.
[11] Volodymyr Babin, Christopher Roland, and Celeste Sagui. Adaptively biased molecular
dynamics for free energy calculations. J. Chem. Phys., 128(13):134101, 2008.
[12] Thomas Huber, Andrew E. Torda, and Wilfred F. van Gunsteren. Local elevation: a
method for improving the searching properties of molecular dynamics simulation. J. Comput.
Aided. Mol. Des., 8:695–708, 1994.
[13] A. Laio and M. Parrinello. Escaping free-energy minima. Proc. Natl. Acad. Sci.,
99:12562–12566, 2002.
[14] M. Iannuzzi, A. Laio, and M. Parrinello. Efficient exploration of reactive potential energy
surfaces using car-parrinello molecular dynamics. Phys. Rev. Lett., 90:238302–1, 2003.


