


Generating a topology: Working with what we have
So far, we have used a number of programs to prepare a very intricate PDB file of the crystal lattice that we want to simulate. The purpose of this approach is really to generate the structure in a file format that is both easily visualized and interpretable by AMBER's preparatory program tLEaP. All that remains, in fact, is to run tLEaP and generate a topology and starting coordinates, then correct the simulation cell dimensions as tLEaP will create an orthorombic box by default.
The following input file for tLEaP is available in XtalUtilities/solvate.tLEaP. It specifies the ff99SB force field, the SPC/E water model, and some custom parameters for ammonium acetate. Numerous bond commands have been specified to unite the sixteen disulfide bridges in the unit cell. The set x box command is used, rather than solvateBox, as we have already solvated the system. As stated above, the box dimensions assigned by tLEaP will need to be specified, and this is done in the input below. The set command command is necessary here to create a topology for a periodic system that will tesselate the unit cell without gaping vacuum regions or clashes. Finally, set default nocenter on command ensures that tLEaP will not change the origin of the coordinates. Later when analyzing the trajectory, this will save us work in finding the optimal location of the origin in order to perform the symmetry operation of the lattice on the asymmetric units.
source leaprc.ff99SB_spce
loadAmberPrep Acetate.prp
loadAmberPrep Ammonium.prp
ammparms = loadAmberParams Ammonium.frcmod
x = loadPdb "solv1AHO.pdb"
bond x.12.SG x.63.SG
bond x.16.SG x.36.SG
bond x.22.SG x.46.SG
bond x.26.SG x.48.SG
bond x.205.SG x.256.SG
bond x.209.SG x.229.SG
bond x.215.SG x.239.SG
bond x.219.SG x.241.SG
bond x.398.SG x.449.SG
bond x.402.SG x.422.SG
bond x.408.SG x.432.SG
bond x.412.SG x.434.SG
bond x.591.SG x.642.SG
bond x.595.SG x.615.SG
bond x.601.SG x.625.SG
bond x.605.SG x.627.SG
set x box {45.900 40.700 30.100}
set default nocenter on
saveAmberParm x solv1AHO.top solv1AHO.crd
quit
Run tLEaP to create the topology and starting coordinates:
[user]$ tleap -f solvate.tleap > solvate.out
As mentioned above, the tLEaP program can impart the correct box lengths by use of the set command. These can be changed manually, but the program ChBox is another way to automate the process. If box angles other than 90 degrees are required, then ChBox is the only utility other than manual intervention to accomplish that change to the initial coordinates.
[user]$ ${AMBERHOME}/bin/ChBox -c solv1AHO.crd -o solv1AHO.crd -X 45.9 -Y 40.7 -Z 30.1 -al 90.0 -bt 90.0 -gm 90.0
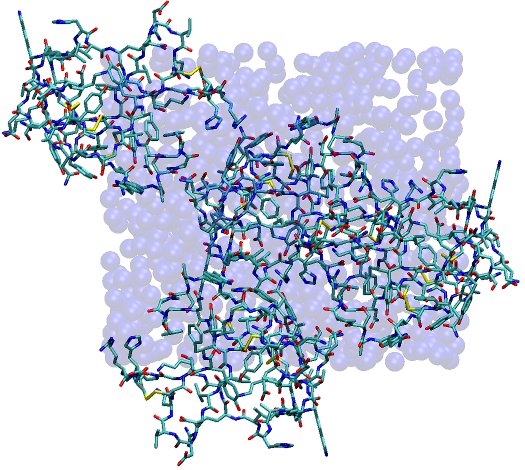
This image depicts the fully solvated unit cell, with water shown as vdW spheres and protein shown in stick form. The boundaries of the unit cell are readily apparent from the distribution of water molecules, which have all been reimaged into the primary unit cell by ptraj. Proteins naturally stick out of the unit cell, but only in such a manner than they fit into spaces left in neighboring unit cells, just as the unit cell depicted contains some spaces (albeit difficult to see in this respresentation).
At this point, the tutorial is essentially over; simulations of this system can now proceed as any typical molecular dynamics calculation, starting from initial coordinates solv1AHO.crd and guided by the topology solv1AHO.top. It is suggested that dynamics utilize a longer cutoff on Lennard-Jones interactions than typical solution phase simulations, and make use of anisotropic box rescaling for pressure corrections (as the system is highly non-isotropic). Here are some suggested run parameters for this system.
Here are a list of references on crystal simulations that may be helpful.
Return to the beginning of the tutorial.