


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Andrew T. Fenley & Michael K. Gilson 2014
Computing Binding Enthalpy Values
Formerly known as AMBER Advanced Tutorial 21: Host-Guest in Explicit Water Example
By Andrew T. Fenley and Michael K. Gilson
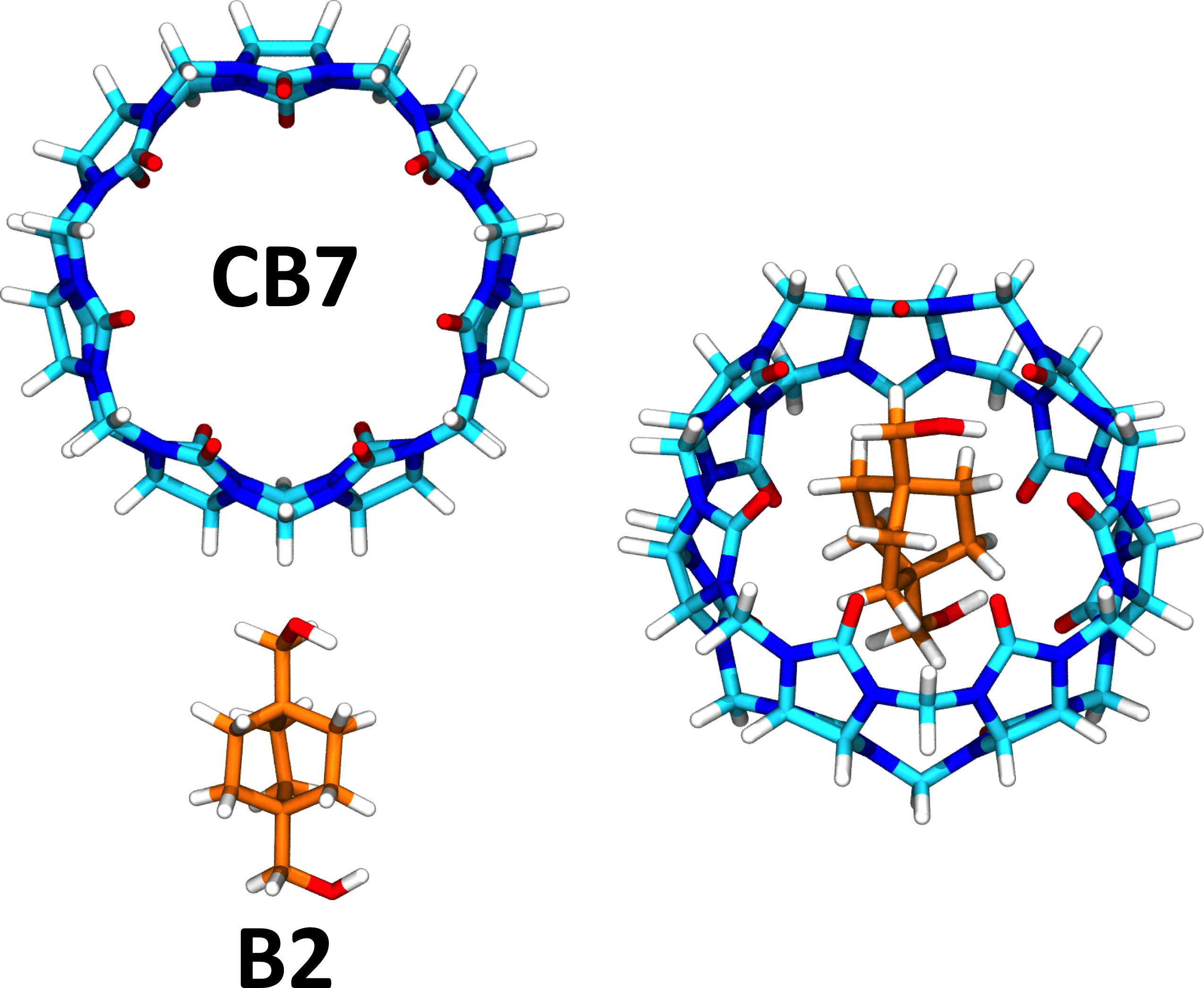
In this tutorial, we will learn how to use the AMBER simulation software package to compute precise binding enthalpies using explicit water molecular dynamics simulations. Here we focus on the guest B2 binding to the host CB7. However, while the example in this tutorial focuses on a host-guest system, the technique is directly applicable to protein-ligand systems although we caution that the such systems will require considerably more sampling to converge than the host-guest system in this tutorial. Additional details regarding the method of computing binding enthalpies are provided in Fenley et al.[1]. This tutorial also assumes familiarity with tleap and the preparation of small molecule structure files with partial charge information. (For additional details see Tutorial 4B.)
Method: The binding enthalpies are computed as follows:
where <H>complex, <H>pure water, <H>host, and <H>guest are the Boltzmann averaged total potential energies for the complex, pure water, host, and guest simulations, respectively. The primary constraint of this calculation is maintaining the same number of atoms per force-field atom type between the bound set of simulations (complex and pure water) and the unbound set of simulations (host and guest). For example, the total number of waters between the bound and unbound sets of simulations must be exactly the same. This is easily achieved for small host-guest systems by setting the number of explicit waters to be the same in all simulations.
The approached described in this tutorial should work for any version of AMBER. However, due to the necessity of computing very long simulations (>500ns), we highly recommend any version of AMBER that supports GPU acceleration (pmemd.cuda). If GPUs are not available, we recommend using pmemd.
This tutorial consists of four sections:
1) section1: Creating the topology and input simulation files.
2) section2: Preparing the system by relaxing bad initial contacts, followed by heating and equilibration stages.
3) section3: Running the production simulations.
4) section4: Computing the binding enthalpy and estimating the uncertainty of the value.
4) section5: Potential pitfalls in computing binding enthalpies.
References
[1] Andrew T. Fenley, Niel M. Henriksen, Hari S. Muddana, and Michael K. Gilson, "Bridging Calorimetry and Simulation Through Precise Calculations of Cucurbituril-Guest Binding Enthalpies", J. Chem. Theory Comput., 2014, in press.
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Andrew T. Fenley & Michael K. Gilson 2014