


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Dwight McGee, Hailey Bureau, Caley Allen, Rigoberto Hernandez
Adaptive Steered Molecular Dynamics
by: T. Dwight McGee Jr., Hailey Bureau, Caley Allen, Rigoberto Hernandez
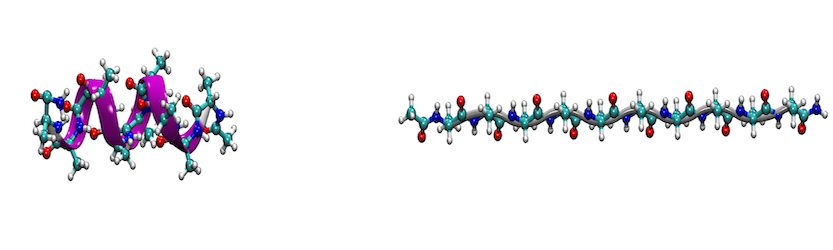
Introduction
In this tutorial, we motivate the use of the AMBER suite to perform adaptive steered molecular dynamics (ASMD) through a calcualtion of the energetics of the peptide, deca-alanine. ASMD was developed by Hernandez and coworkers, and an in-depth description and overview can be found in their articles. 1-5 General familiarity with steered molecular dynamics (SMD)6 will be assumed, as well as familiarity with the AMBER package. This tutorial specifically provides practical steps for performing and analyzing an ASMD simulation.
Brief Background & Theory
SMD utilizes a pseudo particle that applies a steering force in order to tranverse a reaction coordinate at a particular velocity. The external force applied to the system of interest allows one to observe changes in the molecule (in response) within the confines of Molecular Dynamics simulations timescales. Employing the Jarzynski equality7, the non-equilibrium work performed on the system during the SMD simulation can be related to the free energy difference between state A and B.
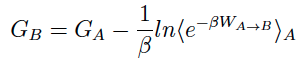
An unfortunate feature underlying the calculation of the exponential-boltzmann-weighted ensemble average is that many simulations must be run in order to converge the Potential Mean of Force (PMF). ASMD has been shown to alleviate this problem. In ASMD, the predetermined reaction coordinate is divided into segments, commonly referred to as stages, within which SMD is performed and the Jarzynski average (JA) is computed throughout the stage see the figure below. In between stages, a single trajectory is selected according to the one whose work value was the closest to the JA average. The coordinates at the end of the stage of the selected trajectory are used to initialize the next stage of SMD trajectories. Contracting the swarm of trajectories to one single JA structure helps to remove trajectories that would make minimum contributions to the overall PMF, thus reducing the number of trajectories that need to be performed.
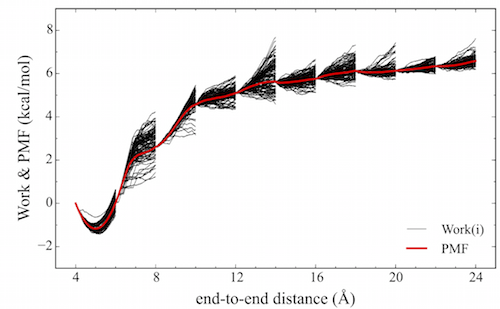
| Schematic of a Potential Mean of Force (red) obtained from an ASMD simulation. Also, highlighted are the work distributions (black) from each stage. |
References
- G. Ozer, S. Quirk, and R. Hernandez, J. Chem. Phys. 136, 215104 (2012).
- G. Ozer, E. Valeev, S. Quirk, and R. Hernandez, J. Chem. Theory Comput. 6, 3026 (2010).
- G. Ozer, S. Quirk, and R. Hernandez, J. Chem. Theory Comput. 8, 4837 (2012).
- G. Ozer, T. Keyes, S. Quirk, and R. Hernandez, J. Chem. Phys. 141, 064101 (2014).
- H. R. Bureau, D. Merz Jr., E. Hershkovits, S. Quirk and R. Hernandez, PLoS ONE 10, e0127034 (2015).
- S. Park and K. Schulten, J. Chem. Phys. 120, 5946 (2004).
- C. Jarzynski Phys. Rev. Lett. 78, 2690 (1997).
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Dwight McGee, Hailey Bureau, Caley Allen, Rigoberto Hernandez