


Copyright (C) Pengfei Li 2016
Setting Up A Polyethylene Terephthalate (PET) Polymer System
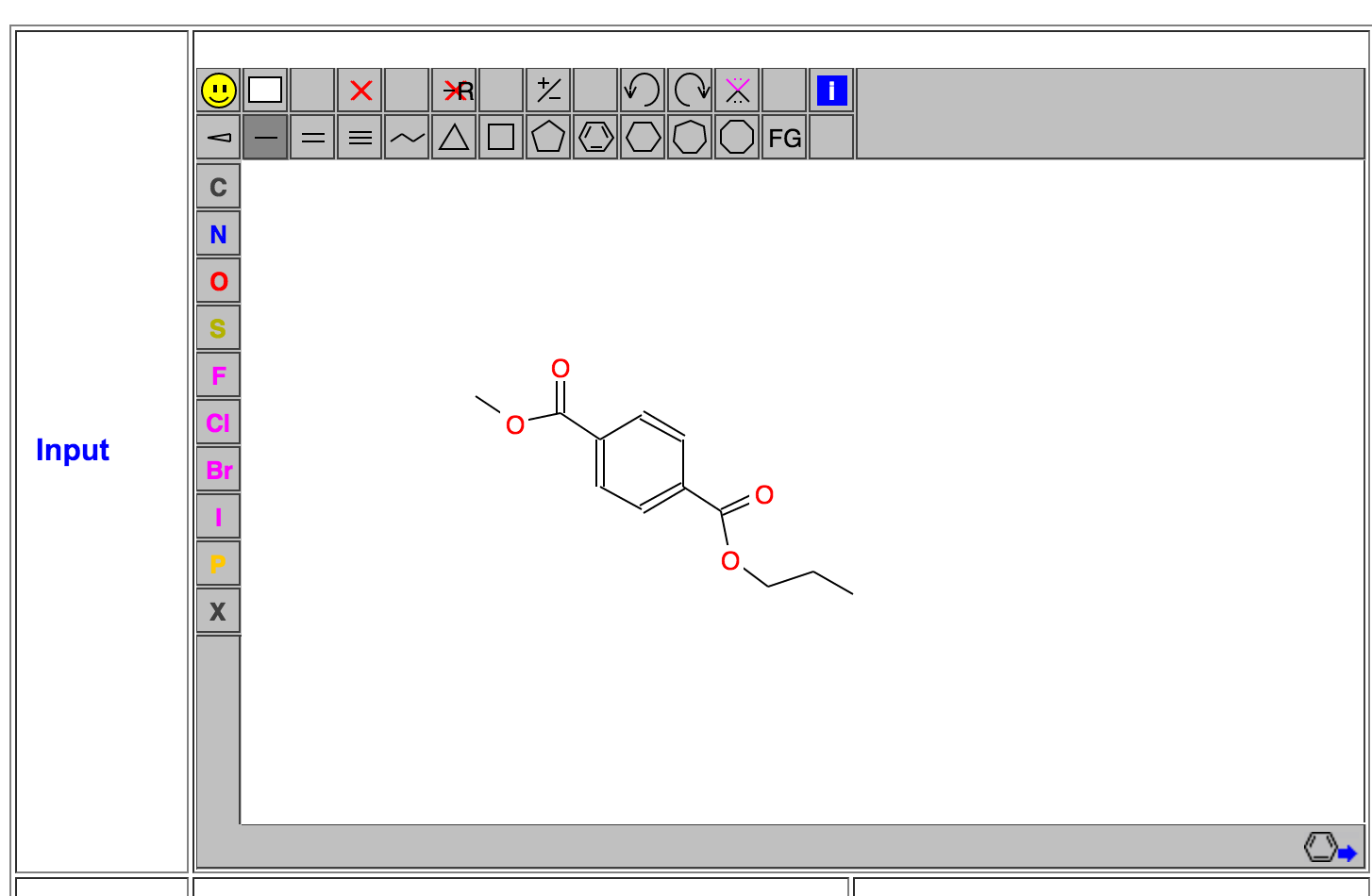
1. Generate a monomer structure:
The PET polymer is introduced in wikipedia: PET. Like to build a force field for protein or DNA, we can build the force field for each monomer inside the polymer system. We need to have a structure for the monomer system at first. Herein we will take advantage of the powerful Graphic antechamber in Prof. Wang's website: Wang's Group. Click "Go to MMFFT"->"Sign In As A Guest"->"Antechamber"->"Graphic Antechamber", then you will see the graphic antechamber, in which you can draw a PET monomer there:
Then we can click the "View 3D Model" button to view the 3D structure:
Meanwhile, there are a lot of options in the "View 3D Model" window you are use here we don't specify. Then you can close the "View 3D Model" window and then click "Run antechamber" in the Graphic antechamber webpage (there are a lot of options you can choose herein we we used the default setting). Then there will be a mol_1_1.mol2 file generated which is for the PET monomer structure. We can right click the file name and download the file through "Save Link As...".
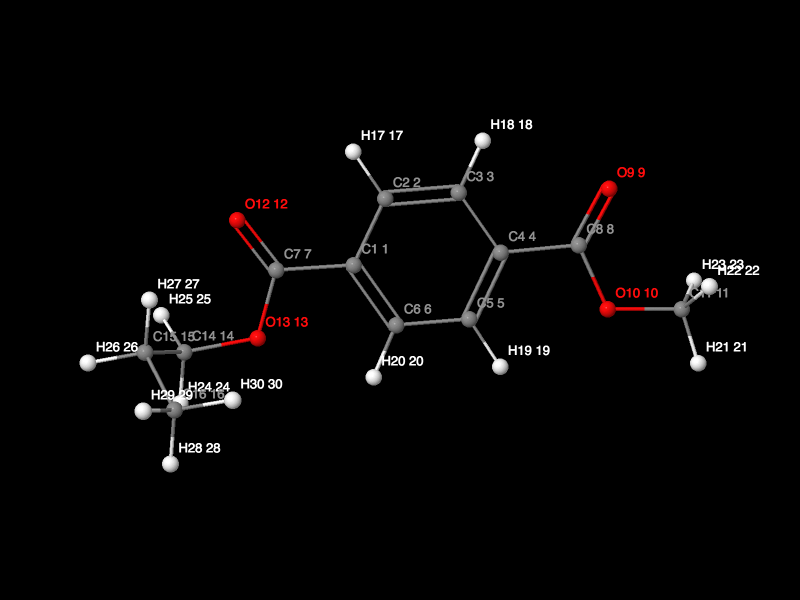
2. Perform Gaussian calculations and do the RESP fits:
After we obtaining the mol2 file, we can use antechamber to transfer the mol2 file to a Gaussian input file for ESP population analysis:
antechamber -fi mol2 -fo gcrt -i mol_1_1.mol2 -o pet.com -pf y
Afterwards we can modify the pet.com a bit and then perform the Gaussian calculation:
g09 < pet.com > pet.log
After the Gaussian calculation finished, the pet.log file store the ESP results, based on which we can perform the RESP fits to generate a ac file (ac is short for antechamber).
antechamber -fi gout -fo ac -i pet.log -o pet.ac -c resp -pf y
3. Build the prepi files for the normal, N-terminal, and C-terminal PET monomers:
We want to have the partial charges for the monomer block in the polymer system, so we need to strip out the charges of the two methyl groups in the monomer structure. The mainchain.pet file is used to define the linkage and strip out information inside the molecule, which needs to be built manually.
prepgen -i pet.ac -o pet.prepi -f prepi -m mainchain.pet -rn PET -rf pet.res
Meanwhile we need to have a head PET residue (like a N-terminal amino acid residue) and a tail PET residue (like a C-terminal amino acid residue) to cap the polymer chain in two ends. Because the terminal PET residues are also neutral, so herein we can perform the partial charge fits based on the same PET ac file, without performing Gaussian calculations specially for the N-terminal and C-terminal PET residues. We can perform the following two commands, in which we used HPT represents the head PET, and TPT represents the tail PET. Again, the mainchain.hpt and mainchain.tpt files are built manually.
prepgen -i pet.ac -o hpt.prepi -f prepi -m mainchain.hpt -rn HPT -rf hpt.res
prepgen -i pet.ac -o tpt.prepi -f prepi -m mainchain.tpt -rn TPT -rf tpt.res
4. Generate a polymer system:
After getting the pet.prepi, hpt.prepi, and tpt.prepi files, we can use them to build a polymer system. The tleap input file is shown here: pet20_tleap.in
Then you can perform the command:
tleap -s -f pet_tleap.in > pet_tleap.out
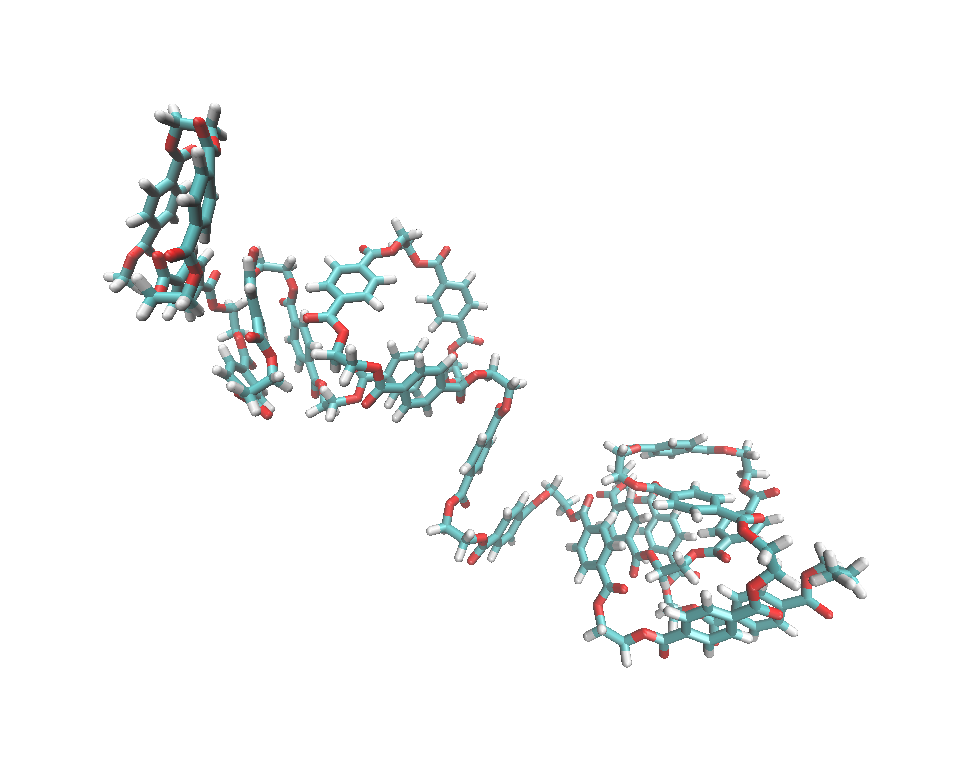
You will get the pet20.prmtop and pet20.inpcrd files for doing simulations.
5. Perform minimization and MD simulations:
Herein we will perform the minimization and MD simulations under gas phase, therefore we need to use sander or sander.MPI to perform these modelings (pmemd and pmemd.cuda are usually only used for condensed phase simulations). To just show an example, in general herein we only performed three steps: minimization, heating, and production. However, practical research may need much more steps to get meaningful data.
Here are the three input files for different stages: pet20_min.in, pet20_heat.in, and pet20_prod.in
And then we can perform the following commands sequentially to perform these simulations:
sander -O -i pet20_min.in -o pet20_min.out -p pet20.prmtop -c pet20.inpcrd -r pet20_min.rst -ref pet20.inpcrd
sander -O -i pet20_heat.in -o pet20_heat.out -p pet20.prmtop -c pet20_min.rst -r pet20_heat.rst -x heat.netcdf -ref pet20_min.rst
sander -O -i pet20_prod.in -o pet20_prod.out -p pet20.prmtop -c pet20_heat.rst -r pet20_prod.rst -x prod.netcdf -ref pet20_heat.rst