


(Note: These tutorials are meant to provide illustrative examples of how to
use AMBER to carry out molecular dynamics simulations. Use them only as a guide
for your own simulations.)
Copyright Jian Yin, Niel M. Henriksen, David R. Slochower & Michael K. Gilson 2016
Computing Binding Free Energy using the Attach-Pull-Release (APR) Method
By Jian Yin, Niel M. Henriksen, David R. Slochower and Michael K. Gilson
Introduction
This tutorial demonstrates how to compute the binding free energies and binding enthalpies of a host-guest system with explicit water using the attach-pull-release (APR) approach and AMBER suite of program. In the APR calculations, molecular dynamics (MD) simulations are used to sample a free energy path in a series of independent windows, in which the substrate is pulled from the binding cavity by imposed restraints. Compared to nonequilibrium pulling techniques such as steered MD, this approach avoids possible energy dissipation which may induce irreversibility. Another big advantage of using independent windows is that simulations can be carried out in parallel on heterogeneous computing architectures. To run the example in this tutorial, work stations that enable GPU acceleration of AMBER are highly recommended. Please remember to set the environment variable "CUDA_VISIBLE_DEVICES" to prevent multiple simulations from running on a single GPU. It is possible to run the tutorial with pmemd.MPI, pmemd, and even sander, but it will take much longer.
The APR protocols have been used to generate moderate to strong correlations between experimental and computational binding thermodynamics based on a broad testing of host-guest systems including cucurbit[7]uril (CB7), octa acid (OA), tetra-endo-methyl octa-acid (TEMOA), α- and β-cyclodextrin (α-CD and β-CD) with guest molecules. For the detailed theoretical framework, methodology, and validation of APR, please refer to the following publications:
Velez-Vega C, Gilson MK. Overcoming Dissipation in the Calculation of Standard Binding Free Energies by Ligand Extraction. J. Comput. Chem., 2013, 34(27), 2360-2371. DOI
Henriksen NM, Fenley AT, Gilson MK. Computational Calorimetry: High-Precision Calculation of Host-Guest Binding Thermodynamics. J. Chem. Theory Comput., 2015, 11(9), 4377-4394. DOI
Yin J, Henriksen NM, Slochower DR, Gilson MK. The SAMPL5 Host-Guest Challenge: Computing Binding Free Energies and Enthalpies from Explicit Solvent Simulations by the Attach-Pull-Release (APR) Method J. Comput. Aided Mol. Des., 2017, 31(1), 133-145. DOI
Fenley AT, Henriksen NM, Muddana HS, Gilson MK. Bridging Calorimetry and Simulation through Precise Calculations of Cucurbituril–Guest Binding Enthalpies. J. Chem. Theory Comput., 2014, 10(9), 4069-4078. DOI
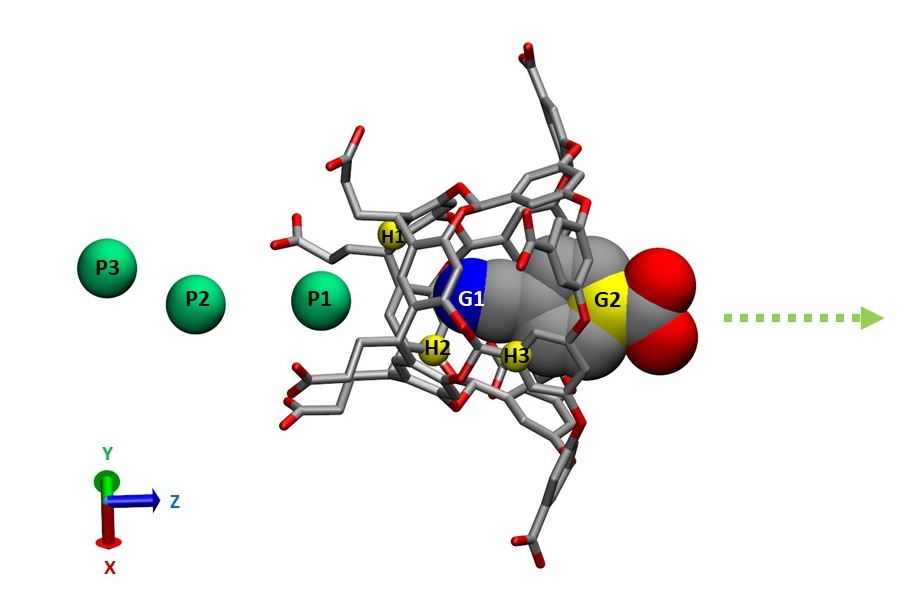
The system of choice here is a host-guest complex: OA and its guest 4-cyanobenzoic acid (also named G2 in the SAMPL5 Challenge). In principle, APR can not only be applied on the relatively simple model systems such as host-guest pairs, but also on more flexible protein-ligand complexes with larger sizes, although careful adjustments of the protocols and scripts will be needed, based on the requirements of every particular system.
This tutorial is organized as below:
Section 1: Prepare the system and set up the restraints.
Section 2: Build the windows and equilibrate the system.
Section 3: Run production phase and analyze data.
Section 4: Compute binding enthalpies within the APR scheme.
Section 5: Adapt the scripts to your own system.