


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright McGee, Miller, and Swails 2009
Python Script MMPBSA.py
Dwight McGee, Bill Miller III, and Jason Swails
1) Set up the pdb files to be leap-ready.
The system we will model in this simulation is the complex between the human H-Ras protein and the Ras-binding domain of C-Raf1 (Ras-Raf), which is central to the signal transduction cascade. Here is a partially equilibrated, pre-prepared pdb file of the RAS-RAF complex.
This structure contains the ras and raf proteins and also a physiologically necessary GTP nucleotide as illustrated in the figure below:

We must now prepare the mutant pdb files to be read by tleap. We strongly suggest preparing this pdb and topology of the files of the mutant along with the initial topology files created in Section 1 prior to running any simulations in order to guarantee consistent prmtop files. For this tutorial we have chosen to mutate residue 21 (Isoleucine, I21) to alanine because this is a residue that is found at the interface between the receptor and ligand and should have a noticeable effect on the binding energy. Note the current version of the code will support mutations to alanine only.
Since I21 is found only in the receptor, we do not need to make a mutant ligand pdb file. Thus, we only need to change ras-raf.pdb and ras.pdb. To do so, you must know something about the structure of the amino acids that are involved. The side chain of isoleucine is -CH(CH3)CH2 CH3. The side chain of alanine is -CH3. Since the side chain of isoleucine has more atoms than the side chain of alanine, we know that we must remove atoms and their corresponding information (name, number, coordinates, etc.) from the pdb files. This mutation involves removing all side chain atoms except the beta-carbon (CB). In I21, this means that we must remove the lines in the Ras-Raf and Ras pdb files corresponding to atoms 294 through 305. We do not need to add the beta-hydrogen (HB) atoms because tleap will add those in the proper locations based on the particular library files you choose for your system. Finally, change the residue name from "ILE" to "ALA" for all remaining atoms of residue 21. This procedure will yield two mutant pdb files: ras-raf_mutant.pdb and ras_mutant.pdb. The I21A mutation in RAS-RAF is depicted below.
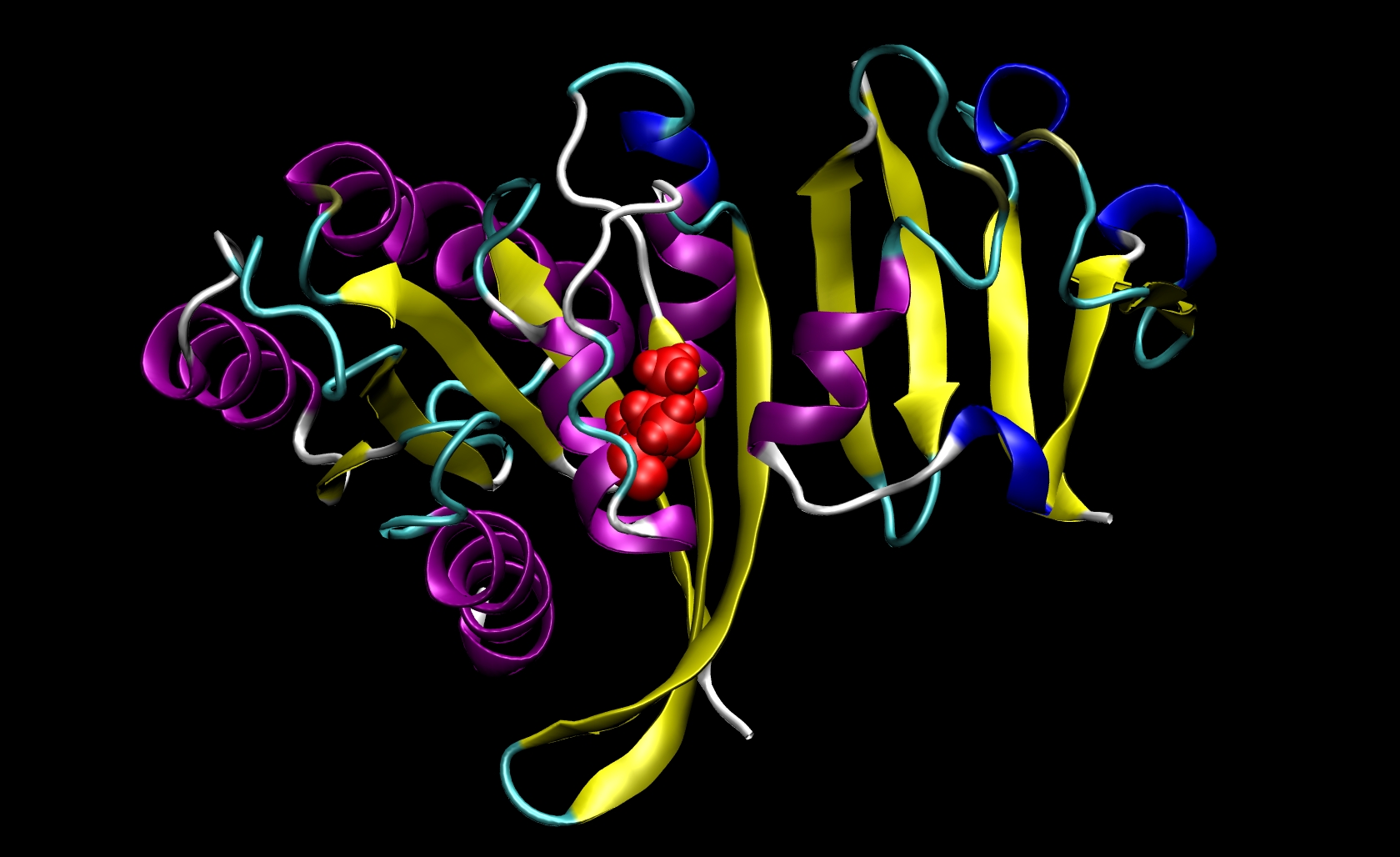
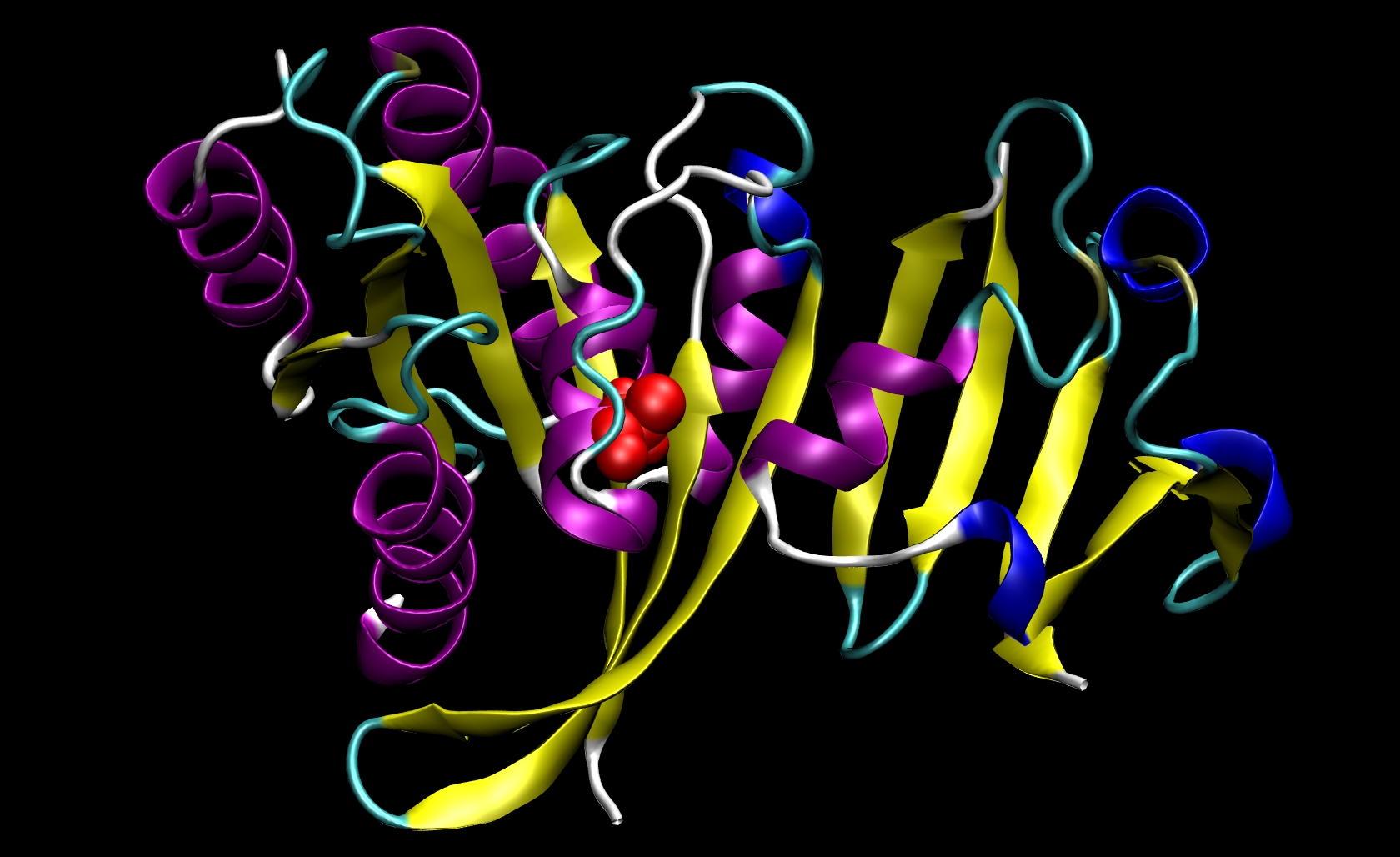
Other mutations will be able to follow similar procedures where the group of atoms after CB but before the carbonyl-carbon (C) may be removed from the pdb file. Note that only one mutation can be performed during a single calculation.
2) Build the starting topology and coordinate files and run a simulation to obtain an equilibrated system.
Now that the pdb files have been made, we need to make the corresponding topology and coordinate files for these structures using tleap. First, we will make the files corresponding to the non-mutant complex:
> $AMBERHOME/exe/tleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99SB
com = loadpdb ras-raf.pdb
ras = loadpdb ras.pdb
raf = loadpdb raf.pdb
saveamberparm com ras-raf.prmtop ras-raf.inpcrd
saveamberparm ras ras.prmtop ras.inpcrd
saveamberparm raf raf.prmtop raf.inpcrd
You should also create the solvated complex for running the MD simulation:
charge com
> Total unperturbed charge: -0.000000
> Total perturbed charge: -0.000000 (Hence there is no need to add counter ions)
solvatebox com TIP3PBOX 12.0
saveamberparm com ras-raf_solvated.prmtop ras-raf_solvated.inpcrd
Now before you quit tleap you should create the topology and coordinate files from the mutant pdb files you just created:
com_mut = loadpdb ras-raf_mutant.pdb
ras_mut = loadpdb ras_mutant.pdb
saveamberparm com_mut rasraf_mutant.prmtop rasraf_mutant.inpcrd
saveamberparm ras_mut ras_mutant.prmtop ras_mutant.inpcrd
quit
We just created 12 files (six .prmtop files and six .inpcrd files). The non-mutant .prmtop and .inpcrd files have been used to run a Molecular Dynamics (MD) simulation to obtain an equilibrated system using the procedures outlined in Section 1 and Section 2.
The important files for calculating the binding free energy using MMPBSA.py are the topology files (non-mutant and mutant) and the mdcrd file ran using the non-mutant topology and coordinate files (ras-raf_alascan.tgz)
3) Perform an alanine scanning calculation on the binding free energy of Ras-Raf.
We will now calculate the interaction energy and solvation free energy for the complex, receptor and ligand and average the results to obtain an estimate of the binding free energy. Then the same calculation will be performed on the mutated structure after the coordinates in the mdcrd file(s) have been mutated for comparison to the "wild-type" structure. Please note that we will not perform a calculation of the entropy contribution to binding in this part of the tutorial and so strictly speaking our result will not be a true free energy but could be used to compare against similar systems. See Section 3.5 for an example of using Normal Mode Analysis (Nmode) to calculate the entropy contribution for a system or uncomment out the last line in the general section to perform a Quasi-Harmonic entropy calculation using the ptraj module in AMBER.
We will carry out the binding energy calculation using both the MM-GBSA method and the MM-PBSA method for comparison. This is accomplished with the following input file for MMPBSA.py:
| mmpbsa.in |
sample input file for running alanine scanning &general startframe=1, endframe=50, interval=1, verbose=1, / &gb saltcon=0.1 / &pb istrng=0.100 / &alanine_scanning / |
The input files for MMPBSA.py are designed to be similar to the setup of an mdin file used in the sander module of AMBER. The start of each namelist is designated by an ampersand (&) followed by the name of the namelist. Furthermore, a backslash (/) or '&end' can be used to end the namelist. For a complete list of all variables please see the Amber Manual. This input file is divided into four namelists: general, pb, gb, and alanine_scanning. The general namelist is designed to specify variables that are not specific to a particular part of the calculation, but rather to all parts. In this setup we have defined RAS to be the receptor and RAF to be the ligand. The 'verbose' variable allows the user to specify what files are removed at the end of the calculation. For more information on the mpi commands, please see the manual or Section 3.4. The '&gb' and '&pb' namelist markers let the script know to perform MM-GBSA and MM-PBSA calculations with the given values defined within those namelists. The 'alanine_scanning' namelist marker initializes alanine scanning in the script. The only recognized input variable in the &alanine_scanning namelist is "mutant_only" which is described in more detail in the manual.
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -sp ras-raf_solvated.prmtop -cp rasraf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd -mc rasraf_mutant.prmtop -mr ras_mutant.prmtop
This will run the script interactively and print the progress of the calculation to STDOUT and any errors or warnings to STDERR. Finally, timings will be printed once the calculation has completed showing the time taken during each step of the calculation.
The '-y *.mdcrd' on the command line tells the script to read in all files in the working directory that end in '.mdcrd' and use them as the trajectories to be analyzed.
Here are all the output files: ALASCAN_output.tgz.
The script creates three unsolvated mdcrd files (complex, receptor, and ligand) using ptraj that are the coordinates analyzed during the GB and PB calculations. The *.mdout files contain the energies for all frames specified. An average pdb file is created as a structure for minimization if entropy calculations are performed. All files created by MMPBSA.py should begin with the prefix '_MMPBSA_' except for the final output file, FINAL_RESULTS_MMPBSA.dat
| FINAL_RESULTS_MMPBSA.dat |
| Run on Thu Feb 11 13:11:48 EST 2010 |Input file: |-------------------------------------------------------------- |sample input file for running alanine scanning | &general | startframe=1, endframe=50, interval=1, | verbose=1, |/ |&gb | saltcon=0.1 |/ |&pb | istrng=0.100 |/ |&alanine_scanning |/ |-------------------------------------------------------------- |Solvated complex topology file: ras-raf_solvated.prmtop |Complex topology file: rasraf.prmtop |Receptor topology file: ras.prmtop |Ligand topology file: raf.prmtop |Initial mdcrd(s): bigprod.mdcrd |Mutant complex topology file: rasraf_mutant.prmtop |Mutant receptor topology file: ras_mutant.prmtop |Mutant ligand topology file: raf.prmtop | |Best guess for receptor mask: ":1-166" |Best guess for ligand mask: ":167-242" |Calculations performed using 50 frames. |Poisson Boltzmann calculations performed using internal PBSA solver in sander. | |All units are reported in kcal/mole. ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- GENERALIZED BORN: Complex: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1863.7944 17.1704 2.4283 EEL -17200.7297 75.9366 10.7391 EGB -3142.2247 63.1977 8.9375 ESURF 91.3565 1.3938 0.1971 G gas -19064.5240 77.8536 11.0102 G solv -3050.8682 63.2131 8.9397 TOTAL -22115.3922 51.5332 7.2879 Receptor: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1268.1888 14.2342 2.0130 EEL -11557.0773 71.7127 10.1417 EGB -2444.8629 54.9156 7.7662 ESURF 64.2843 1.1143 0.1576 G gas -12825.2661 73.1118 10.3396 G solv -2380.5786 54.9269 7.7678 TOTAL -15205.8447 36.8422 5.2103 Ligand: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -529.3090 9.4198 1.3322 EEL -4684.4720 36.1449 5.1117 EGB -1661.8286 26.5442 3.7539 ESURF 37.0493 0.6185 0.0875 G gas -5213.7811 37.3522 5.2824 G solv -1624.7794 26.5514 3.7549 TOTAL -6838.5604 25.6515 3.6277 Differences (Complex - Receptor - Ligand): Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -66.2966 4.2751 0.6046 EEL -959.1803 34.9190 4.9383 EGB 964.4668 32.9201 4.6556 ESURF -9.9770 0.3759 0.0532 DELTA G gas -1025.4769 35.1797 4.9752 DELTA G solv 954.4898 32.9223 4.6559 DELTA G binding = -70.9871 +/- 6.6875 0.9457 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- I21A MUTANT: GENERALIZED BORN: Complex: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1855.4226 17.0765 2.4150 EEL -17210.2882 75.8866 10.7320 EGB -3145.1010 63.2477 8.9446 ESURF 91.8639 1.3913 0.1968 G gas -19065.7108 77.7842 11.0003 G solv -3053.2370 63.2630 8.9467 TOTAL -22118.9478 50.9582 7.2066 Receptor: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1261.9126 14.1817 2.0056 EEL -11566.4419 71.5475 10.1183 EGB -2447.4831 55.0008 7.7783 ESURF 64.5090 1.1105 0.1570 G gas -12828.3545 72.9394 10.3152 G solv -2382.9741 55.0120 7.7799 TOTAL -15211.3287 36.2055 5.1202 Ligand: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -529.3090 9.4198 1.3322 EEL -4684.4720 36.1449 5.1117 EGB -1661.8286 26.5442 3.7539 ESURF 37.0493 0.6185 0.0875 G gas -5213.7811 37.3522 5.2824 G solv -1624.7794 26.5514 3.7549 TOTAL -6838.5604 25.6515 3.6277 Differences (Complex - Receptor - Ligand): Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -64.2010 4.0841 0.5776 EEL -959.3742 34.9114 4.9372 EGB 964.2108 32.9092 4.6541 ESURF -9.6943 0.3800 0.0537 DELTA G gas -1023.5752 35.1495 4.9709 DELTA G solv 954.5164 32.9114 4.6544 DELTA G binding = -69.0588 +/- 6.5302 0.9235 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- RESULT OF ALANINE SCANNING: (I21A MUTANT:) DELTA DELTA G binding = 1.9283 +/- 9.3470 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- POISSON BOLTZMANN: Complex: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1863.7944 17.1704 2.4283 EEL -17200.7297 75.9366 10.7391 EPB -3227.2145 64.4523 9.1149 ECAVITY 68.4754 0.7567 0.1070 G gas -19064.5240 6061.1875 857.1813 G solv -3158.7391 64.4568 9.1156 TOTAL -7522.2032 51.2973 7.2545 Receptor: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1268.1888 14.2342 2.0130 EEL -11557.0773 71.7127 10.1417 EPB -2485.3559 54.5638 7.7165 ECAVITY 47.5088 0.4610 0.0652 G gas -12825.2661 5345.3320 755.9441 G solv -2437.8471 54.5658 7.7168 TOTAL -5118.8075 38.9610 5.5099 Ligand: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -529.3090 9.4198 1.3322 EEL -4684.4720 36.1449 5.1117 EPB -1684.5802 28.2572 3.9962 ECAVITY 28.1687 0.3939 0.0557 G gas -5213.7811 1395.1865 197.3092 G solv -1656.4114 28.2599 3.9966 TOTAL -2313.4381 24.9082 3.5225 Differences (Complex - Receptor - Ligand): Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -66.2966 4.2751 0.6046 EEL -959.1803 34.9190 4.9383 EPB 942.7215 33.8861 4.7922 ECAVITY -7.2022 0.3069 0.0434 DELTA G gas -1025.4769 1237.6138 175.0250 DELTA G solv 935.5194 33.8875 4.7924 DELTA G binding = -89.9575 +/- 8.2480 1.1664 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- I21A MUTANT: POISSON BOLTZMANN: Complex: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1855.4226 17.0765 2.4150 EEL -17210.2882 75.8866 10.7320 EPB -3229.1405 64.8100 9.1655 ECAVITY 68.5521 0.7596 0.1074 G gas -19065.7108 6050.3755 855.6523 G solv -3160.5884 64.8144 9.1661 TOTAL -7520.1586 50.6710 7.1660 Receptor: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -1261.9126 14.1817 2.0056 EEL -11566.4419 71.5475 10.1183 EPB -2487.5603 54.6289 7.7257 ECAVITY 47.6466 0.4632 0.0655 G gas -12828.3545 5320.1609 752.3844 G solv -2439.9137 54.6309 7.7260 TOTAL -5118.8820 38.4370 5.4358 Ligand: Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -529.3090 9.4198 1.3322 EEL -4684.4720 36.1449 5.1117 EPB -1684.5802 28.2572 3.9962 ECAVITY 28.1687 0.3939 0.0557 G gas -5213.7811 1395.1865 197.3092 G solv -1656.4114 28.2599 3.9966 TOTAL -2313.4381 24.9082 3.5225 Differences (Complex - Receptor - Ligand): Energy Component Average Std. Dev. Std. Err. of Mean ------------------------------------------------------------------------------- VDWAALS -64.2010 4.0841 0.5776 EEL -959.3742 34.9114 4.9372 EPB 942.9999 34.0350 4.8133 ECAVITY -7.2632 0.3107 0.0439 DELTA G gas -1023.5752 1235.4872 174.7243 DELTA G solv 935.7367 34.0364 4.8135 DELTA G binding = -87.8385 +/- 8.0665 1.1408 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- RESULT OF ALANINE SCANNING: (I21A MUTANT:) DELTA DELTA G binding = 2.1190 +/- 11.5368 ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- |
The beginning of the statistics file includes the date/time, any warnings based on the values and files given, the mmpbsa.in text, the files used by the script, the number of frames analyzed, and which PB solver (if any) was used. The rest of the statistics file includes all the average energies, standard deviations, and standard error of the mean for GB followed by PB. After each section, the ΔG of binding is given along with the error values. After each method, the ΔΔG of binding is reported that demonstrates the relative affect the mutation has on the ΔG of binding for the complex. The specific mutation is also printed at the end of the file. In this case, we mutated residue 21 from an isoleucine to an alanine (i.e. I21A). The meaning of the different energy terms in this file is as follows:
VDWAALS = van der Waals contribution from MM.
EEL = electrostatic energy as calculated by the MM force field.
EPB/EGB = the electrostatic contribution to the solvation free energy calculated by PB or GB respectively.
ECAVITY = nonpolar contribution to the solvation free energy calculated by an empirical model.
DELTA G binding = final estimated binding free energy calculated from the terms above. (kCal/mol)
Note that the total gas phase energy has not been reported because the values of the bonded potential terms for the receptor and ligand should exactly cancel those for the complex using the single trajectory approach. An error message will result if the energies do not cancel within an allowed tolerance.
One would typically expect to find an extremely favorable electrostatic energy and a unfavorable solvation free energy. This symbolises the energy that ones has to use to de-solvate the binding particles and to align their binding interfaces.
CLICK HERE TO GO TO SECTION 3.4
CLICK HERE TO GO TO MMPBSA.py HOME
CLICK HERE TO GO BACK TO THE INTRODUCTION
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright McGee, Miller, and Swails 2009