


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018
Section 1: Building the initial structure and input files
Preparing the PDB file for LEaP
This section describes how to prepare the input files required for C(pH,E)MD simulations. The first step is to prepare the PDB file to be used in LEaP. As mentioned in Ref. [1], N-acetylmicroperoxidase-8 (NAcMP8) is derived from cytochrome c. The structure of NAcMP8 is sketched in the first figure of Ref. [2]. Based on this figure, we made modifications to the structure of the horse heart cytochrome c, available at the PDB code 1HRC (you can download it here: 1HRC.pdb, or from the PDB website), and we obtained the NAcMP8.pdb file.
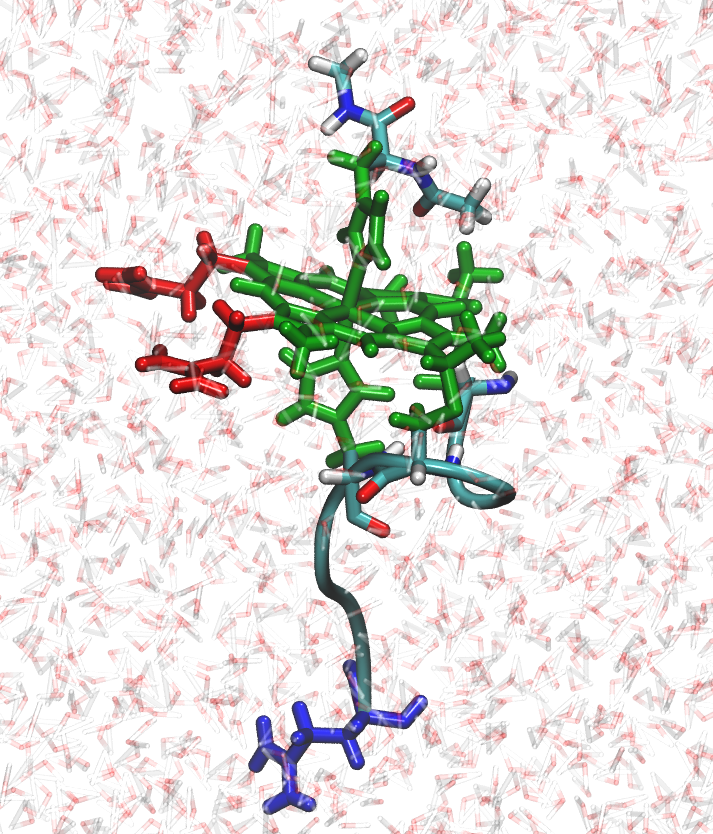 In our leaprc repository prepared for constant Redox Potential simulations (leaprc.conste), the iron atom,
the porphyrin ring, and the side chains of the two histidines and two cysteines were
considered as a single residue, that we call HEH. The HEH residue is colored in green as
shown in the figure at the left and is the redox-active residue that changes its atomic charge
distribution when a reduction state change attempt is accepted. Therefore, when a
redox state change is accepted the charge distribution on the side chains of the
histidines and cysteines are modified as well. NAcMP8 contains three pH-active
residues: two propionates (PRN) colored in red in the figure at the left, and one
glutamate (GL4) colored in blue. The propionates are separate residues from HEH.
In our leaprc repository prepared for constant Redox Potential simulations (leaprc.conste), the iron atom,
the porphyrin ring, and the side chains of the two histidines and two cysteines were
considered as a single residue, that we call HEH. The HEH residue is colored in green as
shown in the figure at the left and is the redox-active residue that changes its atomic charge
distribution when a reduction state change attempt is accepted. Therefore, when a
redox state change is accepted the charge distribution on the side chains of the
histidines and cysteines are modified as well. NAcMP8 contains three pH-active
residues: two propionates (PRN) colored in red in the figure at the left, and one
glutamate (GL4) colored in blue. The propionates are separate residues from HEH.
We now move to a part of the PDB preparation process that requires special attention. Due to the construction of the HEH residue, the atom names of the side chains of the histidines and cysteines that compose our HEH residue have to be unique and match the names defined inside leaprc.conste. The backbone atoms that remain have to be reassigned to new residues we called HIO and CYO (these residues only contain four atoms: N, CA, C and O). Extra care is required here because, for example, the atom names on one of the histidine side chains is not the same as for the other histidine. To make this task easier, in the next table we show the atom names of all HEH atoms (excluding hydrogen atoms). The same is shown for a PRN residue.
Therefore, considering the heme group (HEM) in 1HRC.pdb and NAcMP8.pdb, the first step is to move the atoms of each propionate to independent PRN residues with the atom names shown below. Then, the HEM residue has to be relabeled to HEH, and the histidines and cysteines side chains need to be added to HEH. The atom names on these side chains need to be relabled to match the names in the table below. The cysteines with just backbone atoms need to be relabeled to CYO, and the histidines with just backbone atoms need to be relabeled to HIO. As we will also be titrating the glutamate, we need to relabel GLU→GL4.
The PDB file with the HEH residue correctly set up is then going to be: NAcMP8.constphe.pdb. Use this file as a reference to check if the HEH, PRN, HIO and CYO residues have been correctly placed in your PDB file.
|
|
Executing LEaP
We now need to use tleap to prepare the topology (prmtop) and input coordinate (rst7) files. leaprc.conste contains force field information for HEH, PRN, HIO and CYO. The underlying force field is ff10 (equivalent to ff99SB for proteins). As we also have pH-active residues in our system, we also need to load leaprc.constph. Both leaprc.conste and leaprc.constph sets the appropriate PBRadii set (mbondi2).
We now present leap scripts that will generate the necessary topology and coordinate files for both implicit and explicit solvent simulations:
| tleap.implicit.in | tleap.explicit.in |
|
source leaprc.constph source leaprc.conste # Load PDB mp8 = loadpdb NAcMP8.constphe.pdb # Connecting HEH to the protein bond mp8.14.CB2 mp8.2.CA bond mp8.14.CB1 mp8.9.CA bond mp8.14.CBC1 mp8.8.CA bond mp8.14.CBB2 mp8.5.CA # Connecting HEH to the PRNs bond mp8.14.C3D mp8.16.CA bond mp8.14.C2A mp8.15.CA # Save topology and coordinate files saveamberparm mp8 mp8_is.prmtop mp8_is.rst7 # Quit quit |
source leaprc.constph source leaprc.conste source leaprc.water.tip3p # Load parameters for ions loadAmberParams frcmod.ionsjc_tip3p # Load PDB mp8 = loadpdb NAcMP8.constphe.pdb # Connecting HEH to the protein bond mp8.14.CB2 mp8.2.CA bond mp8.14.CB1 mp8.9.CA bond mp8.14.CBC1 mp8.8.CA bond mp8.14.CBB2 mp8.5.CA # Connecting HEH to the PRNs bond mp8.14.C3D mp8.16.CA bond mp8.14.C2A mp8.15.CA # Solvate solvateOct mp8 TIP3PBOX 10.0 # Add ions addIonsRand mp8 Na+ 2 # Save topology and coordinate files saveamberparm mp8 mp8_es.prmtop mp8_es.rst7 # Quit quit |
|
For implicit solvent simulation the command |
For explicit solvent simulation the command |
(Optional) Script to automatically prepare the PDB file and execute tLeAp
Alternatively to what was presented above, the PDB file for tLeAp, the tLeAp commands (tleap.implicit.in and tleap.explicit.in above), and tLeAp execution can be automatically performed with the aid of a Bash script: SimplifiedSetupForCpHEMD.sh. This script was gracefully prepared by Matthew Guberman-Pfeffer to make this whole process easier. This script requires that you have VMD installed and properly set in your environment. To use the script, simply execute it and follow the prompts on the screen. It will take NAcMP8.pdb as input, and automatically go all the way to generate the mp8_is.prmtop and mp8_is.rst7 or mp8_es.prmtop and mp8_es.rst7 files.
Generating the CPIN and CEIN files
Now we have to generate the constant pH input file (CPIN file) and the constant Redox Potential input file (CEIN file) using both the cpinutil.py and ceinutil.py AmberTools. These files are important because they tell sander or pmemd which residues to be titrated (pH-active and redox-active) during the course of the simulation, and they list the charge distribution of each protonation or redox state for each residue. These files also contain relative energies for each state, and information about the reference pKa or standard Redox Potential (Eo) of each titratable residue.
The Eo for the NAcMP8 axially connected to an imidazole molecule has been experimentally measured as -203 mV vs. NHE at pH 7.0 [2], and this is the reference Eo we use in our simulations for HEH. The experimental pKa of a propionic acid in water is 4.85, and this is the reference pKa used for PRN. The reference pKa of the glutamate residue GL4 is 4.4. You can use the --help flag to obtain a list of available options. (Run cpinutil.py --help or ceinutil.py --help in your terminal).
Our CPIN file will contain information about the pH-active residues (2 PRNs and 1 GL4), and the CEIN file about the redox-active residue HEH. By default, all protonation and redox states start at state 0. For PRN and GL4 this means deprotonated, and for HEH this means oxidized. You may run cpinutil.py --describe or ceinutil.py --describe in your terminal to see these definitions for other residues. We will generate our CPIN and CEIN files with the default starting states.
| Implicit Solvent: | Explicit Solvent: | |
|
The following command
And the following command |
The following command
And the following command The new prmtop file is necessary because it contains a correction to fix the radii for the carboxylate oxygens of any AS4, GL4 or PRN residues you wish to titrate. Because residues that contain carboxylate groups are defined with 4 hydrogens (one in the syn- and another in the anti- positions for each carboxylate oxygen), the effective Born radius is artificially increased as a result of the extra hydrogen atoms. To compensate that, the radii is decreased in the prmtop file. |
We now have all the files necessary to run a constant pH and Redox Potential MD simulation. It is always a good idea to visualize your system at this point to make sure that everything is correct, including all the bonds HEH makes with the PRN, CIO and HIO residues.
Note: if you want to run simulations for redox-active residues that are not yet parametrized, please check the section Extending constant Redox Potential to additional titratable groups in the Amber manual. The finddgref.py AmberTool or TI calculations can be used to compute the necessary reference energies of each redox state.
Click here to go back to the Introduction
References
[1] Vinícius Wilian D. Cruzeiro, Marcos S. Amaral, and Adrian E. Roitberg, "Redox Potential Replica Exchange Molecular Dynamics at Constant pH in AMBER: Implementation and Validation", J. Chem. Phys., 149, 072338 (2018). DOI: https://doi.org/10.1063/1.5027379
[2] H.M. Marques, I. Cukrowski, and P.R. Vashi, "Co-ordination of weak field ligands by N-acetylmicroperoxidase-8 (NAcMP8), a ferric haempeptide from cytochrome c, and the influence of the axial ligand on the reduction potential of complexes of NAcMP8", J. Chem. Soc. Dalt. Trans., 8, 1335 (2000).
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018