Placing Customized Extra Points in an Amber Topology File
The mdgx &parmedit module serves a purpose with overlap in
parmed and tleap, and while those programs could, in time, take
over this functionality it was expedient to have the feature in mdgx as there is
no file format precedent for it. Extra points, or "virtual sites" are a powerful way to
improve the monopole distribution of a molecule. The mdgx program has the ability
to fit partial charges for these extra points once they are part of the topology. Prior to
AmberTools21, the extra points had to be added as "post-translational modifications" to the
topology, only mdgx could read them, and mdgx could only add them at runtime (no topology, and
hence no coordinate set containing the new points, could be produced). With the addition of
topology writing capabilities and a new format for encoding the extra points, these features of
the system can start to become more permanent. The current master branch of pmemd
and pmemd.cuda support the upgraded topology format, and is available in Amber22. Users can experiment with adding extra points,
fitting partial
charges or bonded
parameters using the associated mdgx modules, and run short simulations
with the new models using the mdgx CPU PME dynamics capability.
Topologies are still made with tleap, but can now be modified using a new file
format that makes use of the mdgx &rule namelist. Like all other
mdgx namelists, the list of keywords for &rule and
&parmedit can be obtained by running:
mdgx -RULE
mdgx -PARMEDIT
on the command line. The &parmedit module merely takes directives for
the names of the new topology and coordiante files along with an optional topology title and
requests to check the total charge and connectivity. The &rule namelist is
much more detailed, allowing users to set all non-bonded aspects of existing as well as new,
massless particles. The ability to modify existing particles (including atoms with mass) is
crucial to conserve charges on a system if a new extra point is added. Seven other, unique
"frame" types may be specified, denoting how the extra point will be positioned according to
the locations of atoms that do have mass.
For the first example, we will convert TIP3P water into TIP4P-Ew water. Because the TIP3P
water molecule is rigid, there are several frame types that would produce equivalent results,
and we will choose the simplest of them. The following &rule file will guide
the conversion:
&rule
ExtraPoint 'EPW',
ResidueName 'WAT',
FrameType 'Flex-3',
ParentAtom 'O',
FrameAtom2 'H1',
FrameAtom3 'H2',
Vector12 0.106641378,
Vector13 0.106641378,
Charge -1.04844
&end
&rule
ResidueName 'WAT',
FrameType 'None',
AtomName 'O',
Charge 0.0,
Sigma 3.164349,
Epsilon 0.162750,
&end
&rule
ResidueName 'WAT',
FrameType 'None',
AtomName 'H1',
Charge 0.52422,
&end
&rule
ResidueName 'WAT',
FrameType 'None',
AtomName 'H2',
Charge 0.52422,
&end
This input, taking the &rule namelist file to be "tip4p.rules", will then
convert any topology containing TIP3P waters:
&files
-p tip3p.prmtop
-c tip3p.inpcrd
-xpt tip4p.rules
&end
&parmedit
NewPrmtop tip4pew.prmtop,
NewInpcrd tip4pew.inpcrd,
&end
Running the above input as "convert.mdin" with this
topology file and these
input coordinates produces a new
topology file and
coordinates wherein all of the waters have been converted
to TIP4P-Ew parameters. Note that, while this is a handy trick, it is for demonstrative
purposes. There is no advantage to changing the water model in this way rather than loading
TIP4P-Ew from the start. Furthermore, there is no way to modify the names of atoms or residues
using &rule namelists, if these are important for later analysis. The new
topology contains some new fields towards the bottom:
%FLAG VIRTUAL_SITE_FRAMES
%FORMAT(10I8)
4 1 2 3 -1 5 8 5 6 7
-1 5 12 9 10 11 -1 5 16 13
(...)
2444 2441 2442 2443 -1 5 2448 2445 2446 2447
-1 5
%FLAG VIRTUAL_SITE_FRAME_DETAILS
%FORMAT(5E16.8)
1.06641378E-01 1.06641378E-01 0.00000000E+00 1.06641378E-01 1.06641378E-01
0.00000000E+00 1.06641378E-01 1.06641378E-01 0.00000000E+00 1.06641378E-01
(...)
1.06641378E-01 1.06641378E-01 0.00000000E+00 1.06641378E-01 1.06641378E-01
0.00000000E+00
These new sections will direct pmemd, pmemd.cuda, and
mdgx to see the relevant connections between the new, massless atoms and their
frames. The VIRTUAL_SITE_FRAMES section contains one tuple of six integers per extra point.
They describe the index of the new particle, the indices of up to four frame atoms (marked -1
if there is no second, third, or fourth frame atom), and the frame type index (an internal
numbering system dictated by the string found after the obligatory "FrameType" keyword in each
&rule). The VIRTUAL_SITE_FRAME_DETAILS dection contains tuples of three reals for every
frame, marked with zeros if any of the three numbers is not relevant.
As mentioned before, were the TIP4P-Ew water model to be loaded from the beginning, these
new topology segments would not be present and all of the Amber programs would default to their
original behavior of inferring the extra point types by seeing atoms with no mass and then
looking at connectivities recorded as actual bonds between each extra point and one other atom.
Also, note that tip4pew.prmtop contains Lennard-Jones coefficients for three atom types whereas
tip3p.prmtop contains coefficients only for two. There are Lennard-Jones ACOEF and BCOEF
values for the interactions of TIP3P and TIP4P-Ew oxygen atoms, even though such a mixed system
would probably never be built. mdgx has given the re-parameterized oxygen atom a
new type index (3), and since all of the oxygens used to be of type 1 there simply are no
longer any atoms of this type. The new extra point, like the hydrogens, has no Lennard-Jones
properties and takes type index 2, just like them. The two topologies do not work the same
way, and the custom extra point format will not work in sander as of yet, but in
our faster engines the two topologies will create water with identical properties.
It is also helpful to check the resulting mdout. The topology
checking features are engaged by default, and they report that all of the residues in TIP4P-Ew
water still have neutral net charges. A total of 612 water molecules received an extra
point.
It is more common to want to add extra points to a specific ligand or side chain. The next
example will place extra points on a bromobenzene molecule near the bromine sigma hole. It
begins with a much more extensive set of &rule
namelists. One of the weaknesses of this process is that mdgx will often
report "atom not found" if there is a misstatement in the rule namelists. It is therefore
helpful to construct the file one namelist at a time to ensure that such a mistake is
immediately corrected. Aside from that, mdgx prints many helpful messages to
guide the user in stating the input in the best possible syntax.
The associated &parmedit input follows
from the previous example. The new topology is not that
large, as there is only one residue to apply the extra point onto. Here, the extra points get
their own atom type because there is no polar hydrogen, no atom with no Lennard-Jones
properties. The first extra point is placed using just two frame atoms, at a fixed distance of
-0.8 Angstroms from the bromine along the bromine-carbon bond. Subsequent extra points are
placed in the benzene ring's Π-system. As in the previous example, the
new coordinate file provides a means for visualizing the
result:
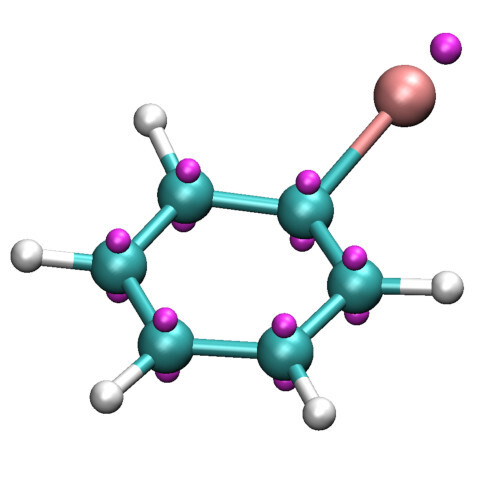
The mdout file again provides some useful information for keeping track of what was added.
Unique extra points are identified by unique frame geometries, but extra points with similar
geometries and different frame atoms are grouped together for the purposes of this summary.
Each extra point is shown beside its parent atom.
Extra points were added to this topology.
Frame Type Type Alias Inherited Added
---------- ------------ --------- ---------
Out-3 OutOfPlane 0 12
FD-2 FixedDis2 0 1
Details of added extra points follow.
Example format:
[Extra Point] --
[Parent Atom] --
Virtual Site ID 0 ( 1 instances):
Frame type: FD-2 / FixedDis2
Charge: -0.25000
LJ-Sigma: 0.00000
LJ-Epsilon: 0.00000
Example: [ LIG 0 -- SH1 12 ] -> [ LIG 0 -- Br1 11 ]
Virtual Site ID 0 ( 6 instances):
Frame type: Out-3 / OutOfPlane
Charge: -0.10000
LJ-Sigma: 0.00000
LJ-Epsilon: 0.00000
Examples: [ LIG 0 -- PL1A 13 ] -> [ LIG 0 -- C1 4 ]
Virtual Site ID 0 ( 6 instances):
Frame type: Out-3 / OutOfPlane
Charge: -0.10000
LJ-Sigma: 0.00000
LJ-Epsilon: 0.00000
Examples: [ LIG 0 -- PL1B 14 ] -> [ LIG 0 -- C1 4 ]
There are many other frame types that mdgx can impart to a topology and that
pmemd or pmemd.cuda can then run at the best speed of any Amber
simulation. While it is not ideal to have this topology modification uniquely supported by
mdgx, the combination of topology and coordinate manipulations needed to make this
work was a natural extension of features already in the program. Fitting parameters for models
with these extra points is its own challenge, and will be elaborated in a subsequent tutorial.
However, mdgx has all of the tools to make this work.
|


