


Hydrogen Mass Repartitioning
Implimentation of methods found in:Hopkins, C.W., Le Grand, S.L., Walker, R.C. and A.E. Roitberg, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J. Chem. Theory Comput., 2015, 11(4), 1864-1874.
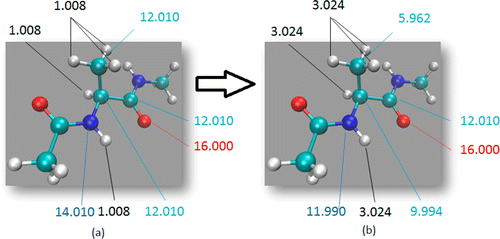
Figure 1: Hydrogen Mass Repartitioning example with Alanine Dipeptide (a) Dialanine peptide atomic masses. (b) Dialanine peptide masses after repartitioning. Notice that the overall mass remains the same.
Learning Outcomes
Introduction
Hydrogen mass repartitioning (HMR) is very useful because it allows a larger time step to be used for your simulation by redistributing some of the mass from heavy atoms connected to hydrogen into the bonded hydrogens. This means that simulations can accurately represent a longer time frame without encountering instability-related errors caused by high-frequency hydrogen motion. Some biological studies are interested in large motions of biomolecules. These require longer simulation times, which can be accomplished by using hydrogen mass repartitioning. For instance, if one wanted to run a simulation with a 4 fs time step instead of a more customary 2 fs time step, then one could use parmed (below) to perform hydrogen mass repartitioning on an existing parm7 topology file and then run equilibration and MD.For more information about hydrogen mass repartitioning, please read Roitberg and coworkers .
Process
1. Use parmed to load a topology file built in LEaPFor this tutorial, we will use the topology file made during the 5.1 Simple Simulation of Alanine Dipeptide tutorial.
The alanine dipeptide parm7 file is available here:
diala.parm7
parmed diala.parm7
ParmEd: a Parameter file Editor
Loaded Amber topology file parm7
Reading input from STDIN...
>
This command both triples the mass of all hydrogens on alanine dipeptide and scales down the mass of all other atoms on alanine dipeptide, preserving the total mass (See Figure 1). To learn more about the hmassrepartition command, please look through the HMassRepartition subsection on page 271 of the Amber 2021 Manual.
hmassrepartition
Repartitioning hydrogen masses to 3.024 daltons. Not changing water hydrogen masses.
>
Give this file a new name that says "hmass" so you know it was modified. Close parmed with the quit command.
outparm diala_hmass.parm7
Outputting Amber topology file diala_hmass.parm7
> quit
Done!
The new file hmass.parm7 should look like this diala_hmass.parm7
By Jan Ziembicki, Carlos Simmerling and Maria Nagan