


Preparing PDB files with pdb4amber
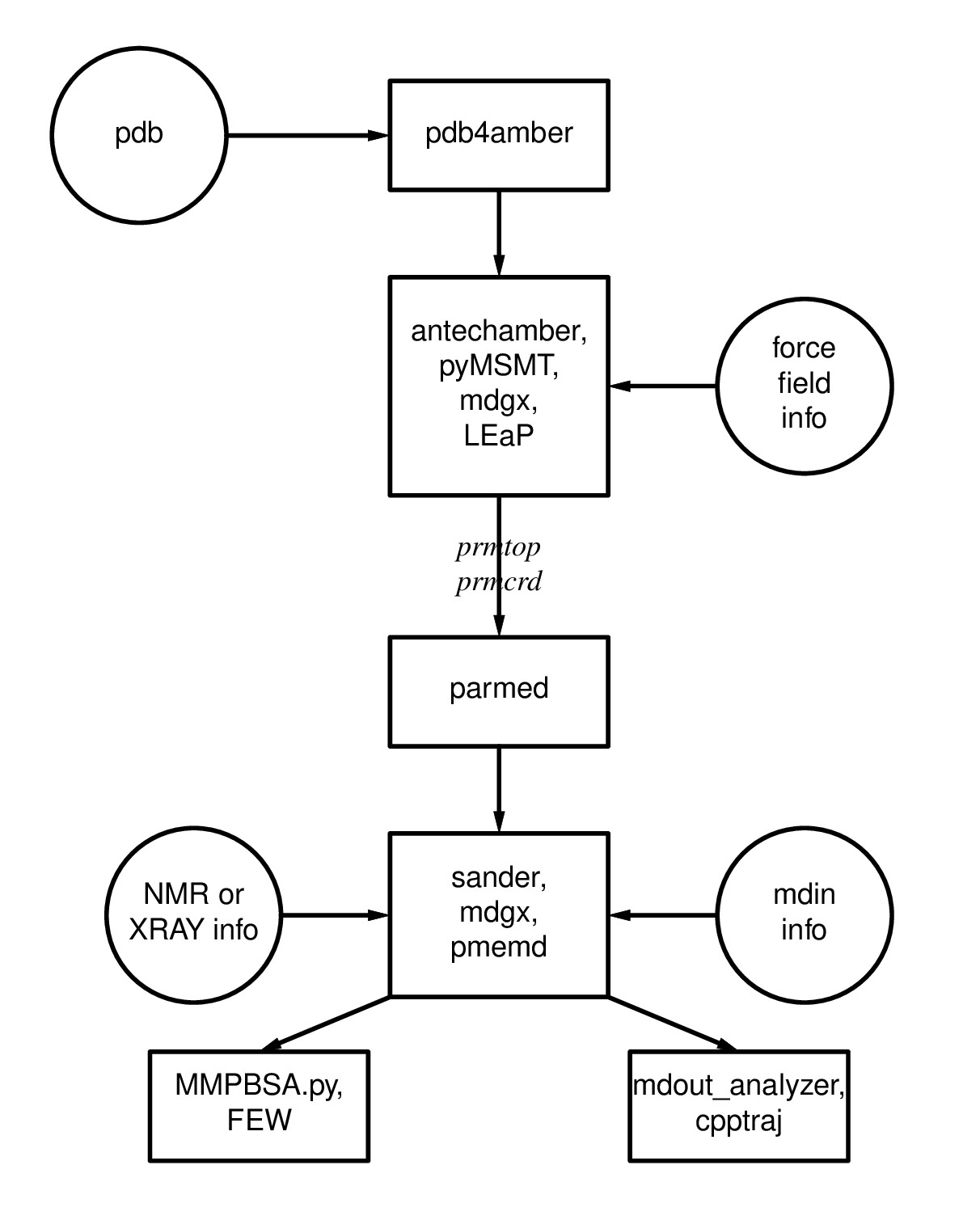 Figure 1.1: Basic information flow of Amber
Figure 1.1: Basic information flow of Amber Learning Outcomes
Introduction
This is not really a tutorial, but instead a page meant to highlight the existence of pdb4amber. It used to be a little difficult to load pdb files into LEaP, especially RNA molecules, which sometimes use disparate naming conventions in the pdb files. Proteins also have missing sections, disulfide bonds and different HIS protonation states, to name a few of the issues. pdb4amber highlights these issues, which require more careful attention, as is pointed out in Tutorial 1.4 Building Protein Systems in Explicit Solvent. pdb4amber analyses and cleans PDB files for further usage, especially within the LEaP programs of Amber. To learn more about the pdb4amber command, please look through section 13.4 of the Amber Manual.
To get a sense for the capabilities of pdb4amber, type
%pdb4amber -h
usage: pdb4amber [-h] [-i FILE] [-o FILE] [-y] [-d] [-s STRIP_ATOM_MASK]
[-m MUTATION_STRING] [-p] [-a] [--constantph]
[--most-populous] [--keep-altlocs] [--reduce]
[--no-reduce-db] [--pdbid] [--add-missing-atoms]
[--model MODEL] [-l FILE] [-v] [--leap-template]
[--no-conect] [--noter]
[input]
positional arguments:
input PDB input file (default: stdin)
optional arguments:
-h, --help show this help message and exit
-i FILE, --in FILE PDB input file (default: stdin)
-o FILE, --out FILE PDB output file (default: stdout)
-y, --nohyd remove all hydrogen atoms (default: no)
-d, --dry remove all water molecules (default: no)
-s STRIP_ATOM_MASK, --strip STRIP_ATOM_MASK
Strip given atom mask, (default: no)
-m MUTATION_STRING, --mutate MUTATION_STRING
Mutate residue
-p, --prot keep only protein residues (default: no)
-a, --amber-compatible-residues
keep only Amber-compatible residues (default: no)
--constantph rename GLU,ASP,HIS for constant pH simulation
--most-populous keep most populous alt. conf.
(default is to keep 'A')
--keep-altlocs Keep alternative conformations
--reduce Run Reduce first to add hydrogens.
(default: no)
--no-reduce-db If reduce is on, skip using it for hetatoms.
(default:usual reduce behavior for hetatoms)
--pdbid fetch structure with given pdbid, should be
combined with -i option. Subjected to change
--add-missing-atoms Use tleap to add missing atoms.
(EXPERIMENTAL OPTION)
--model MODEL Model to use from a multi-model pdb file (integer).
(default: use 1st model). Use a negative number to
keep all models
-l FILE, --logfile FILE
log filename
-v, --version version
--leap-template write a leap template for easy adaption
(EXPERIMENTAL)
--no-conect Not write S-S conect record
--noter Not writing TER
Process
1. Download a PDB fileWe will use the RCSB Protein Databank to download 1ESH, the NMR structure of a small RNA triloop from Brome Mosaic Virus. Go to the pdb databank and type the PDBID into the search bar.

Click on the actual entry. You will see a blue Download Files icon. Click on it and select the pdb file.

The pdb file contains only the lowest energy structure of 1ESH. It still has its header information on it.
2. Convert the PDB file to Amber format using pdb4amber> pdb4amber 1esh.pdb > 1esh.amber.pdb
=====================================================
Summary of pdb4amber for: 1esh.pdb
=====================================================
----------Chains
The following (original) chains have been found:
A
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
---------- Mising heavy atom(s)
None
>
Click on the filename to download the processed file 1esh.amber.pdb.
Conclusion
Having completed the first 2 steps of Figure 1.1, you are ready to make topology (prmtop) and coordinate (inpcrd) files using LEaP. For more information on topology and coordinate files as well as the flow of information in Amber, please read through section 1.1 of the Amber Manual.To do so, you will have to load a force field which describes the potential energy of the molecules in your system. Different force fields are used for different types of molecules (e.g., DNA, RNA, proteins, lipids, carbohydrates). To learn more about force fields, please refer to section 3 of the Amber Manual.
For more information about building systems with LEaP, go back to the Building Systems tutorial page and find information relevant for your system.
By Jan Ziembicki and Maria Nagan