


Simulating a Lipid Bilayer Tutorial

TABLE OF CONTENTS
- Introduction
- Lipid14
- Lipid Bilayer Structures
- CHARMM-GUI
- Lipid14 PDB Format
- LEaP
- Molecular Dynamics
- Analysis with cpptraj
- Membrane-Bound Proteins
- Additional Resources
- References
Introduction
Lipids are a crucial component of lipid bilayers and play an important role in many cell signaling and physiological processes. Condensed phase molecular dynamics software packages like Amber are now able to simulate a variety of biomolecules, including lipids.This Tutorial
In this tutorial, we present a step-by-step guide to setting up a lipid bilayer system and running molecular dynamics with Amber and the Lipid14 force field. We assume that you are using a Unix system with Amber14 successfully installed. We also assume you have run basic Amber molecular dynamics simulations before.This tutorial shows how to simulate a lipid bilayer and a lipid bilayer with a membrane-bound protein. It is often important to understand the dynamics of the bilayer itself before proceeding with protein systems. This tutorial goes on to provide details of how to set up membrane bound proteins systems using Amber and Lipid14.
Warning
This tutorial describes running lipid molecular dynamics simulations for instructional purposes. The settings in this tutorial are designed for this tutorial and will need to be adapted to other systems. A detailed understanding of each step will help to make informed decisions for other systems.
Prerequisites
- Amber and AmberTools version 14.0 installed on a Unix based machine (Linux of Mac OS).
- Lipid14 force field
Lipid14 is included in AmberTools 14 by default. - charmmlipid2amber.py
This is included in AmberTools 14 update 1. - VMD 1.9.1
A molecular visualization program.
Lipid14
Please cite your use of the Lipid14 force field with:Dickson, C.J., Madej, B.D., Skjevik, A.A., Betz, R.M., Teigen, K., Gould, I.R., Walker, R.C., "Lipid14: The Amber Lipid Force Field", J. Chem. Theory Comput., 2014, 10(2), 865-879.
DOI: 10.1021/ct4010307
Amber 14 includes Lipid14 [1], a modular lipid force for tensionless lipid phospholipid simulations. Lipid14 includes the modular charge derivation framework developed in Lipid11 [2] as well as a reparameterization of key van der Waals and dihedral angles as performed in GAFFlipid [3]. Lipid14 has been extensively tested and validated on six key lipid bilayer types. Lipid14's parameterization strategy is consistent and compatible with the approach taken by other pairwise-additive Amber force fields. Therefore, Lipid14 is, in principle, fully compatible with the other biomolecular force fields included in Amber.
The currently supported lipid "residues" are listed below as well as the validated lipid bilayers. For reference, we also list the previous lipids in GAFFlipid and Lipid14. Additional head groups and tail groups as well as other lipid bilayer components such as cholesterol will be included in upcoming updates of the Lipid14 force field.
Lipid14 Residues
| Description | LIPID14 Residue Name | |
| Acyl Chain | Lauroyl (12:0) Myristoyl (14:0) Palmitoyl (16:0) Stearoyl (18:0) Oleoyl (18:1 n-9) | LA MY PA ST OL |
| Head Group | Phosphatidylcholine Phosphatidylethanolamine | PC PE |
Lipid11 Residues
| Description | LIPID11 Residue Name | |
| Acyl Chain | Palmitoyl (16:0) Stearoyl (18:0) Oleoyl (18:1 n-9) Linoleoyl (18:2 n-6) Linolenoyl (18:3 n-3) Arachidonoyl (20:4 n-6) Docosahexanoyl (22:6 n-3)) | PA ST OL LEO LEN AR DHA |
| Head Group | Phosphatidylcholine Phosphatidylethanolamine Phosphatidylserine Phosphatidic acid (PHO4 -) Phosphatidic acid (PO4 2-) R-phosphatidylglycerol S-phosphatidylglycerol | PC PE PS PH- P2- PGR PGS |
| Other | Phosphatidylinositol Cholesterol | PI CHL |
GAFFlipid Molecules
| Name |
GAFFlipid Molecule Name |
| 1,2-dilauroyl-sn-glycero-3-phosphocholine 1,2-dimyristoyl-sn-glycero-3-phosphocholine 1,2-dioleoyl-sn-glycero-3-phosphocholine 1,2-dipalmitoyl-sn-glycero-3-phosphocholine 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
DLPC DMPC DOPC DPPC POPC POPE |
Lipid Bilayer Structures
In order to simulate a lipid bilayer, it is necessary to first obtain a starting structure in PDB format that can be processed by Amber's LEaP program. There are several accessible options for building a lipid structure:- CHARMM-GUI Lipid Builder
A simple, internet based solution to generating lipid bilayer structures as well as membrane-bound protein structures
- VMD Membrane Builder plugin
A plugin is available for the Visual Molecular Dynamics (VMD) program that can currently build POPC and POPE bilayer systems. A tutorial explaining how to build lipid bilayers as well as insert membrane-bound proteins is available on the VMD web site.
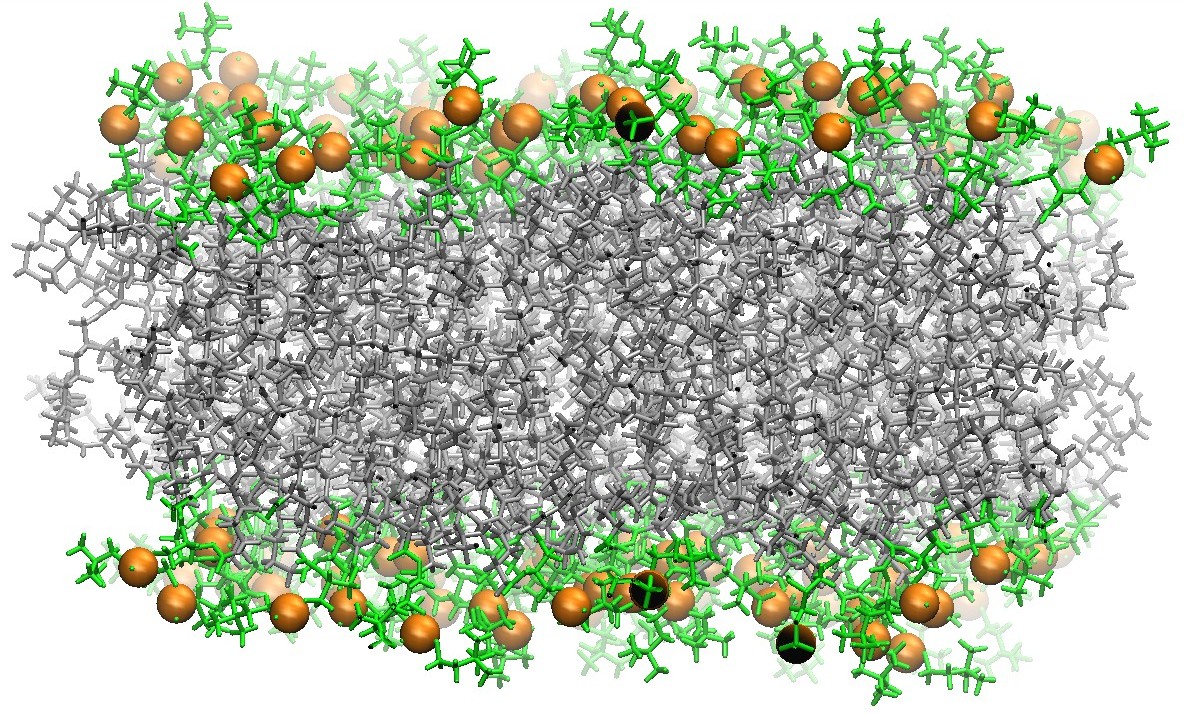
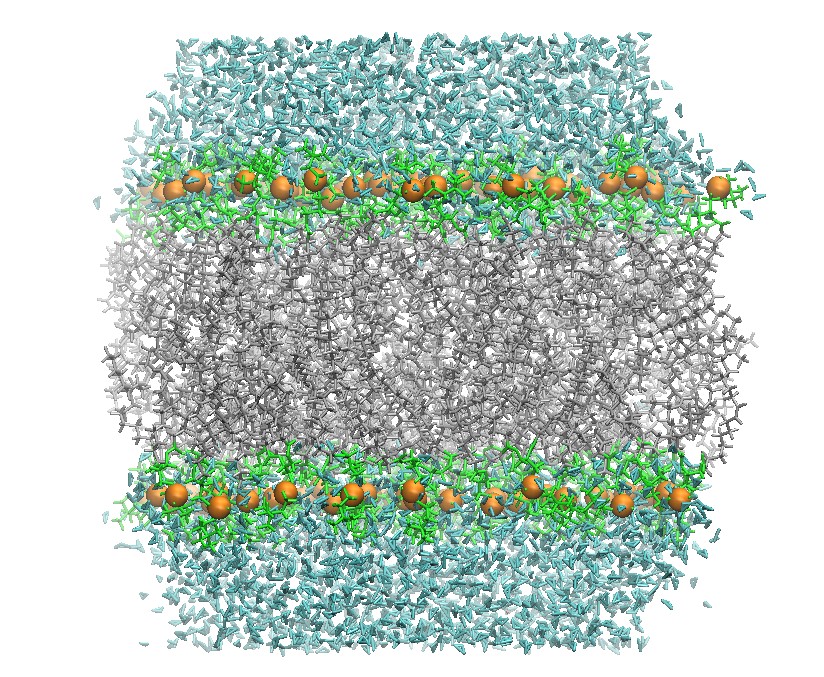
128 DOPC Phospholipid Bilayer Cross-Section
CHARMM-GUI
Go to the CHARMM-GUI Lipid Builder
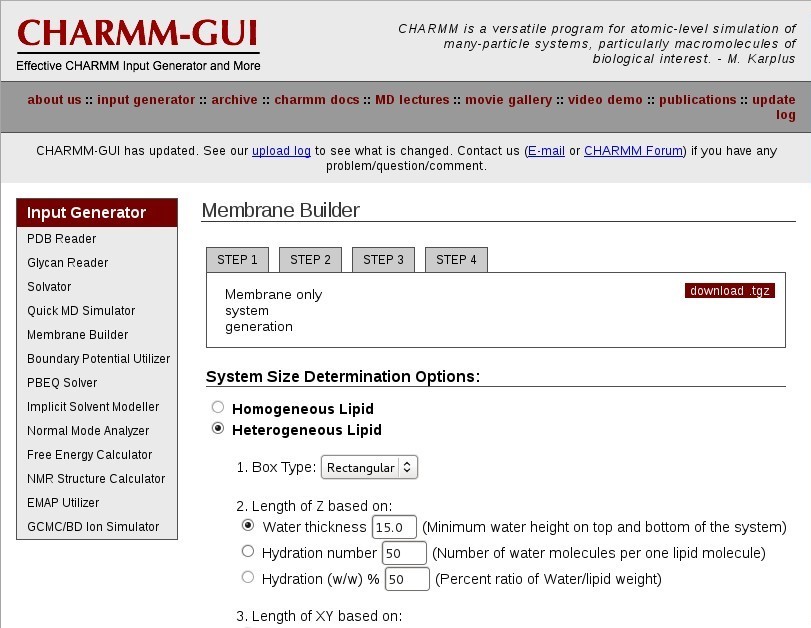
Membrane Builder
Choose "Membrane Only System".
System Size Determination
Choose a "Heterogeneous Lipid" system.
Your system will be rectangular and periodic in the X, Y, and Z dimensions. You will be simulating a patch of bilayer with periodic boundary conditions.
Choose a "Rectangular" box type.
Choose an appropriate hydration number for your lipid type to define the box Z length. This is important for a properly solvated bilayer system. It is suggested that you review the literature for appropriate hydration numbers for various lipids.
For DOPC, use "37" waters per lipid, in excess of the experimental 32.8 waters per lipid [4].
Select "Numbers of lipid components' for your system to define the box XY length. This will create a system based on the surface area per lipid.
Now enter the "# of Lipid on Upperleaflet" and "# of Lipid on Lowerleaflet". See the CHARMM-GUI table below for a list of currently supported CHARMM-GUI lipids.
For DOPC, set "64" on the lower leaflet and "64" on the upper leaflet.
Click "Show the system info" and verify the system properties are correct.
Continue to the next step.
Component Building Options
Depending on your lipid type and interfacial environment, you will want to include an ion concentration. It is important to include ions due to the inherent charge of the lipid head groups and their interaction with water solvent. Some systems may have special ionic concentration requirements: for example in protein ion channel systems.
For DOPC, set the ion concentration at "0.15" M "KCl". Choose the "Monte-Carlo" ion placing method. Continue to the next step.
Building Ion and Waterbox
The next two steps build the lipid bilayer system into a single structure file.
Continue to the next step.
Assemble Generated Components
Continue to the next step.
Determine System Size and Equilibration Options
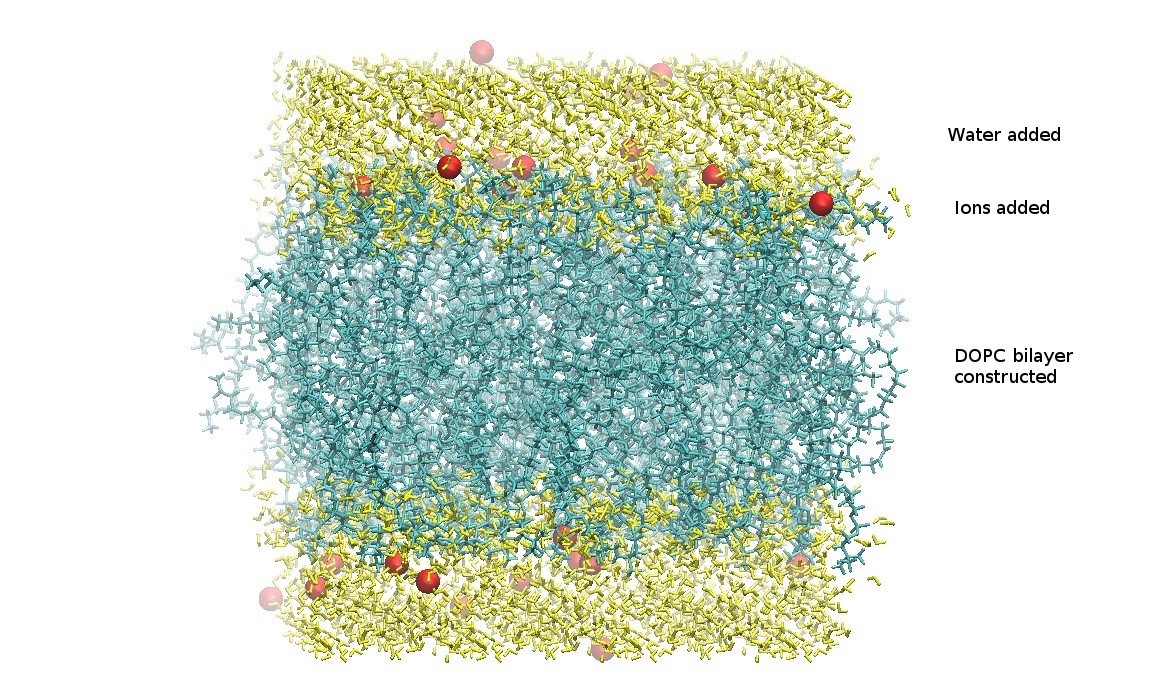
At this point, the generated lipid, waters, and ions have been combined into a single structure PDB file.
Download the .tgz file and save it to your computer.
step5_assembly.pdb
From the charmm-gui.tgz file, the only file needed is the assembled PDB file step5_assembly.pdb. This file should have the lipid bilayer, the water molecules, and included ions. It is important to verify all chosen components are present.
Extract step5_assembly.pdb and save to your working directory.
CHARMM-GUI Construction
Currently Supported CHARMM-GUI Lipids with Lipid14
*Can be built with VMD builder and processed with charmmlipid2amber.py
| CHARMM-GUI Lipid | Lipid14 Residue 1 | Lipid14 Residue 2 | Lipid14 Residue 3 |
| DLPC DMPC DPPC DOPC POPC* | LA MY PA OL PA | PC PC PC PC PC | LA MY PA OL OL |
| DLPE DMPE DPPE DOPE POPE* | LA MY PA OL PA | PE PE PE PE PE | LA MY PA OL OL |
Lipid14 PDB Format
Now you have an initial lipid bilayer PDB file. However, there is no standard for structuring lipid PDB files.Briefly, the Lipid14 format is as follows: The lipid is split into three "residues": two tail groups and one head group. Each residue in Lipid14 has a specific residue name, atom names, and atom types. In a Lipid14 PDB file, a phospholipid molecule is listed as a chain of three lipid residues in specific order: sn-1 tail group, head, sn-2 tail group. Each molecule consisting of a chain of three residues must be ended with a TER card.
Lipid14 PDB File Format
# Phospholipid 1The CHARMM-GUI PDB formatted file must be reformatted to file that follows the Lipid14 format that LEaP expects.
Acyl chain 1 residue
Head residue
Acly chain 2 residue
TER card
# Phospholipid 2
Acyl chain 1 residue
Head residue
Acly chain 2 residue
TER card
...
charmmlipid2amber.py
charmmlipid2amber.pycharmmlipid2amber.py is a robust residue and atom renaming and reordering script available with AmberTools 14 update 1. It is used here to convert the CHARMM-GUI PDB format structures into a PDB format that can be read with LEaP and Lipid14. If you do not have AmberTools 14 update 1 available, you can download the zip file from this web site. This can be placed in your Amber directory and unzipped there.
Warning
charmmlipid2amber.py has replaced the charmmlipid2amber.x script. It is included in AmberTools 14 update 1.
Now, to convert the CHARMM-GUI formatted PDB file to the Lipid14 formatted PDB file, use the script charmmlipid2amber.py.
charmmlipid2amber.py usage:
charmmlipid2amber.py -i input_structure.pdb \input_structure.pdb is the CHARMM-GUI formatted PDB file and output_structure.pdb is the Lipid14 formatted PDB file to be loaded with LEaP.
[-c substitution_definitions.csv] -o output_structure.pdb
For the 128 lipid DOPC system, convert the PDB file:
$ charmmlipid2amber.py -i step5_assembly.pdb \
-c $AMBERHOME/AmberTools/src/etc/charmmlipid2amber/charmmlipid2amber.csv \
-o DOPC_128.pdb
After the script completes processing the PDB file, you should have a file DOPC_128.pdb ready to be loaded into LEaP.
Estimate Periodic Box Size
Due to the nature in which CHARMM-GUI builds lipid bilayers, there will be lipids extending beyond the solvation layers in the X and Y dimensions. It is possible to better estimate the periodic box dimensions by using the water molecules' coordinates for this measurement. This can be manually measured using a PDB structure viewer like VMD.However, we provide a simple bash/VMD script here to measure the periodic box dimensions here:
vmd_box_dims.sh
vmd_box_dims.sh usage:vmd_box_dims.sh -i input_structure.pdb -s vmd_selectionNow calculate your box dimensions from the water molecules:
$ vmd_box_dims.sh -i DOPC_128.pdb -s water
81.35599899291992 80.00199890136719 70.54999923706055
LEaP
In this section, you will load the file named DOPC_128.pdb into LEaP to define the topology and parameters for the lipid residues making up each phospholipid. Use this method to prepare an Amber parameter topology file and initial coordinate file.Start xLEaP:
$ xleapIn the LEaP command line, source the force fields you want to use. Lipid14 is designed to be compatible with the other pairwise-additive Amber force fields.
Load the force fields:
> source leaprc.lipid14With the processed file, DOPC_128.pdb, LEaP will load the structure, split the lipids into three units, and assign atom types.
> source leaprc.ff12SB
> loadamberparams frcmod.ionsjc_tip3p
Load the lipid structure file into a unit:
> DOPC = loadpdb DOPC_128.pdbNote
There will be warnings about lipids being split into different residues. This is normal LEaP behavior. The residue sequence number may not correspond to the residues in your PDB file.
Now we use the periodic box size as measured from the water molecules in our structure using VMD.
Add the periodic box to the system:
> set DOPC box { 81.35599899291992 80.00199890136719 70.54999923706055 }
Save the Amber prmtop parameter and topology file and the inpcrd initial coordinate file for the molecular dynamics simulation:> saveAmberParm DOPC DOPC_128.prmtop DOPC_128.inpcrdNote
Pay close attention to any error messages that LEaP provides when saving the parameter and topology file. Verify that there are no missing parameters.
DOPC_128.prmtop
DOPC_128.inpcrd
At this point the necessary files have been generated to run the actual simulation.
Quit LEaP and verify that the prmtop and inpcrd files were created.
> quit
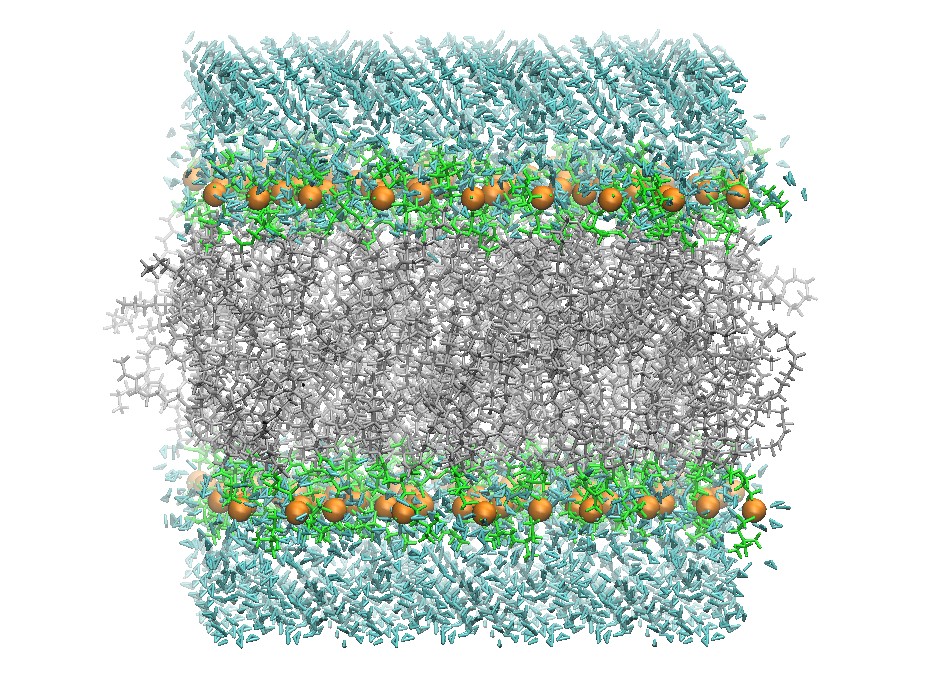
Initial DOPC 128 Lipid Bilayer
Molecular Dynamics
WarningMolecular dynamics simulations of lipid bilayers is not trivial. It is suggested that you first review the literature regarding lipid bilayer simulations before proceeding. The method presented below is similar to that used in the Lipid14 publication.
For molecular dynamics of lipid bilayer systems the following protocol can be used:
- Minimization
- Heating, holding the lipids fixed
- Heating 2, holding the lipids fixed
- 10X Hold, to equilibrate periodic box dimensions
- Production with constant pressure
Minimization
The first step is essential to minimize and relax the initially generated structure. This method of minimization is fairly common in Amber molecular dynamics simulations.Note
The input files below are commented to provide an explanation of the various options used
01_Min.in
Lipid minimizationIn order to run the minimization with pmemd, use the following command:
&cntrl
imin=1, ! Minimize the initial structure
maxcyc=10000, ! Maximum number of cycles for minimization
ncyc=5000, ! Switch from steepest descent to conjugate gradient
minimization after ncyc cycles
ntb=1, ! Constant volume
ntp=0, ! No pressure scaling
ntf=1, ! Complete force evaluation
ntc=1, ! No SHAKE
ntpr=50, ! Print to mdout every ntpr steps
ntwr=2000, ! Write a restart file every ntwr steps
cut=10.0, ! Nonbonded cutoff in Angstroms
/
$ pmemd -O -i 01_Min.in -o 01_Min.out -p DOPC_128.prmtop \After the minimization is complete, you can read the mdout file for the details of the minimization. The important file for the next stage of simulation is the 01_Min.rst restart file, which will be used for the heating portion of the molecular dynamics.
-c DOPC_128.inpcrd -r 01_Min.rst &
128 DOPC Lipid Bilayer After Minimization

Heating
After the initial minimization, slowly heat the system to production temperature. Choosing a production temperature is an important choice in the lipid bilayer simulation. Lipid bilayers have experimentally-measured phase transition temperatures from highly ordered gel-like phases to liquid phases. One major problem in lipid bilayer simulations is accurately simulating the phase transition of lipids.For DOPC, a production temperature of 303 K is acceptable because it is well above the phase transition temperature. The system was heated through two sequential runs to 303 K while keeping the lipid fixed. First the system is heated to 100 K and then slowly to the production temperature. Use the following input for the first heating step:
02_Heat.in
Lipid 128 heating 100KNote that the Langevin thermostat is used for the initial heating. In addition, the nmropt=1 allows one to vary the system temperature throughout the heating. The lipid molecules are restrained using a harmonic restraint and the GROUP input section at the end of the input file.
&cntrl
imin=0, ! Molecular dynamics
ntx=1, ! Positions read formatted with no initial velocities
irest=0, ! No restart
ntc=2, ! SHAKE on for bonds with hydrogen
ntf=2, ! No force evaluation for bonds with hydrogen
tol=0.0000001, ! SHAKE tolerance
nstlim=2500, ! Number of MD steps
ntt=3, ! Langevin thermostat
gamma_ln=1.0, ! Collision frequency for Langevin thermostat
ntr=1, ! Restrain atoms using a harmonic potential
! (See the GROUP input below)
ig=-1, ! Random seed for Langevin thermostat
ntpr=100,
ntwr=10000,
ntwx=100, ! Write to trajectory file every ntwx steps
dt=0.002, ! Timestep (ps)
nmropt=1, ! NMR restraints will be read (See TEMP0 control below)
ntb=1,
ntp=0,
cut=10.0,
ioutfm=1, ! Write a binary (netcdf) trajectory
ntxo=2, ! Write binary restart files
/
&wt
type='TEMP0', ! Varies the target temperature TEMP0
istep1=0, ! Initial step
istep2=2500, ! Final step
value1=0.0, ! Initial temp0 (K)
value2=100.0 / ! final temp0 (K)
&wt type='END' / ! End of varying conditions
Hold lipid fixed
10.0 ! Force constant (kcal/(mol Angstroms^2))
RES 1 384 ! Choose residues
END
END ! End GROUP input
To run the first heating moleculary dynamics, use the following command:
$ pmemd.cuda -O -i 02_Heat.in -o 02_heat.out -p DOPC_128.prmtop \Unfortunately because of the size of the trajectory files, this tutorial web site cannot host these files.
-c 01_Min.rst -r 02_Heat.rst -ref 01_Min.rst -x 02_Heat.nc
Heating 2
The second phase of heating slowly increases the temperature to the desired production temperature. The positions and velocities are read from the previous restart file. This time an anisotropic Berendsen weak-coupling barostat is used to also equilibrate the pressure in addition to the use of the Langevin thermostat to equilibrate the temperature.03_Heat2.in
Lipid 128 heating 303KTo run the second heating molecular dynamics, use the following command:
&cntrl
imin=0,
ntx=5, ! Positions and velocities read formatted
irest=1, ! Restart calculation
ntc=2,
ntf=2,
tol=0.0000001,
nstlim=50000, ! Number of MD steps
ntt=3,
gamma_ln=1.0,
ntr=1,
ig=-1,
ntpr=100,
ntwr=10000,
ntwx=100,
dt=0.002,
nmropt=1,
ntb=2, ! Constant pressure periodic boundary conditions
ntp=2, ! Anisotropic pressure coupling
taup=2.0, ! Pressure relaxation time (ps)
cut=10.0,
ioutfm=1,
ntxo=2,
/
&wt
type='TEMP0',
istep1=0,
istep2=50000,
value1=100.0,
value2=303.0 /
&wt type='END' /
Hold lipid fixed
10.0
RES 1 384
END
END
$ pmemd -O -i 03_Heat2.in -o 03_Heat2.out -p DOPC_128.prmtop \
-c 02_Heat.rst -r 03_Heat.rst -ref 02_Heat.rst -x 03_Heat.nc
128 DOPC Lipid Bilayer after Heating
Hold
In order to equilibrate the system's periodic boundary condition dimensions, it is necessary if you are using the GPU code pmemd.cuda, to run 5ns MD with a barostat. The system's dimensions and density must equilibrate before proceeding with production MD. Because the periodic boundary condition box dimensions are changing, it is necessary to increase the "skinnb" value and to restart the MD simulation after 500ns. This should avoid most "skinnb" errors.The reason for this is that, for performance reasons, the GPU code does not recalculate the non-bond list cells during a simulation. If these cells change size, due to the box changing size, by too much then it will cause the code to halt with an error related to skinnb. Once the system is equibrated the box size fluctuations should be small and so this should not be an issue during production.
04_Hold.in
Lipid production 303K 500psWe run this hold production 10 times (5ns) to equilibrate the system before production MD. This can be scripted easily.
&cntrl
imin=0,
ntx=5,
irest=1,
ntc=2,
ntf=2,
tol=0.0000001,
nstlim=250000,
ntt=3,
gamma_ln=1.0,
temp0=303.0,
ntpr=5000,
ntwr=5000,
ntwx=5000,
dt=0.002,
ig=-1,
ntb=2,
ntp=2,
cut=10.0,
ioutfm=1,
ntxo=2,
/
/
&ewald
skinnb=5, ! Increase skinnb to avoid skinnb errors
/
To run production of the lipid bilayer, use the following command and run the following:
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_1.out -p DOPC_128.prmtop \
-c 03_Heat2.rst -r 04_Hold_1.rst -x 04_Hold_1.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_2.out -p DOPC_128.prmtop \
-c 04_Hold_1.rst -r 04_Hold_2.rst -x 04_Hold_2.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_3.out -p DOPC_128.prmtop \
-c 04_Hold_2.rst -r 04_Hold_3.rst -x 04_Hold_3.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_4.out -p DOPC_128.prmtop \
-c 04_Hold_3.rst -r 04_Hold_4.rst -x 04_Hold_4.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_5.out -p DOPC_128.prmtop \
-c 04_Hold_4.rst -r 04_Hold_5.rst -x 04_Hold_5.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_6.out -p DOPC_128.prmtop \
-c 04_Hold_5.rst -r 04_Hold_6.rst -x 04_Hold_6.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_7.out -p DOPC_128.prmtop \
-c 04_Hold_6.rst -r 04_Hold_7.rst -x 04_Hold_7.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_8.out -p DOPC_128.prmtop \
-c 04_Hold_7.rst -r 04_Hold_8.rst -x 04_Hold_8.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_9.out -p DOPC_128.prmtop \
-c 04_Hold_8.rst -r 04_Hold_9.rst -x 04_Hold_9.nc
$ pmemd.cuda -O -i 04_Hold.in -o 04_Hold_10.out -p DOPC_128.prmtop \
-c 04_Hold_9.rst -r 04_Hold_10.rst -x 04_Hold_10.nc
Production
Actual production molecular dynamics can be run with the input file shown below. Temperature is controlled here using the Langevin thermostat while pressure is controlled using the anisotropic Berendsen barostat.Use this input file for the production of lipid bilayers:
05_Prod.in
Lipid production 303K 125nsTo run production of the lipid bilayer, use the following command:
&cntrl
imin=0, ! Molecular dynamics
ntx=5, ! Positions and velocities read formatted
irest=1, ! Restart calculation
ntc=2, ! SHAKE on for bonds with hydrogen
ntf=2, ! No force evaluation for bonds with hydrogen
tol=0.0000001, ! SHAKE tolerance
nstlim=62500000, ! Number of MD steps
ntt=3, ! Langevin thermostat
gamma_ln=1.0, ! Collision frequency for thermostat
temp0=303.0, ! Simulation temperature (K)
ntpr=5000, ! Print to mdout every ntpr steps
ntwr=500000, ! Write a restart file every ntwr steps
ntwx=5000, ! Write to trajectory file every ntwc steps
dt=0.002, ! Timestep (ps)
ig=-1, ! Random seed for Langevin thermostat
ntb=2, ! Constant pressure periodic boundary conditions
ntp=2, ! Anisotropic pressure coupling
cut=10.0, ! Nonbonded cutoff (Angstroms)
ioutfm=1, ! Write binary NetCDF trajectory
ntxo=2, ! Write binary restart file
/
$ pmemd.cuda -O -i 05_Prod.in -o 05_Prod.out -p DOPC_128.prmtop \
-c 04_Hold_10.rst -inf -r 05_Prod.rst -x 05_Prod.nc
128 DOPC Lipid Bilayer after 5 ns Production Molecular Dynamics

Using the restart files and this same input file this simulation can be continued until the structure has converged. In particular, it is common to continue production until the bilayer and the structure's area per lipid has converged.
Analysis with cpptraj
After production, there are several lipid bilayer parameters worth
examining including area per lipid, electron density profiles,
deuterium order parameters, as well as others. Current lipid molecular
dynamics literature present many methods of analysis. This is an
introduction to several of the techniques.There are a variety of scripts available to examine some of the lipid bilayer properties. As an introduction to analysis of the lipid bilayer properties, this section explains how to obtain the area per lipid and time-averaged electron density profile for a trajectory.
Area per Lipid
Area per lipid is the average area that a single phospholipid occupies in an interface and is usually reported in Angstroms squared. It can be calculated with:Area per lipid = (box X dimension) * (box Y dimension) / (number of phospholipids per layer)
cpptraj is an Amber trajectory analysis program that can perform a variety of actions on trajectory files. In order to calculate the area per lipid of a lipid bilayer system, you can use this program to extract the periodic box dimensions from a trajectory. In order to run cpptraj, an input file is needed for this operation. A sample input file with comments is provided below.
box_dimension.cpptraj
# Read in trajectory fileIn order to run cpptraj on the trajectory use the command:
trajin 04_Hold_1.nc
trajin 04_Hold_2.nc
trajin 04_Hold_3.nc
trajin 04_Hold_4.nc
trajin 04_Hold_5.nc
trajin 04_Hold_6.nc
trajin 04_Hold_7.nc
trajin 04_Hold_8.nc
trajin 04_Hold_9.nc
trajin 04_Hold_10.nc
trajin 05_Prod.nc
# Write the vector named "test" (selecting all atoms)
# of box coordinates out to a file named "vector.dat"
vector test * box out vector.dat
cpptraj -i box_dimension.cpptraj -p DOPC_128.prmtop
The file vector.dat has the box dimensions stored in columns in the file. However the first two lines of the file is a header that must be removed for the calculation.
Use tail to strip the first two lines of the file:
tail -n +3 vector.dat > vector_2.dat
To actually calculate the area per lipid as a function of time, the box x dimension (column 2) must be multiplied by the box y dimension (column 3).
Use awk to do the arithmetic on vector2.tmp:
awk '{print ($2 * $3) / 64}' vector_2.dat > area_per_lipid.dat
area_per_lipid.dat can be easily plotted with your favorite plotting tool.
For example, to plot with gnuplot, start gnuplot:
$ gnuplot
Plot with the gnuplot:
> plot "area_per_lipid.dat" with lines
Area per Lipid of 128 DOPC Bilayer
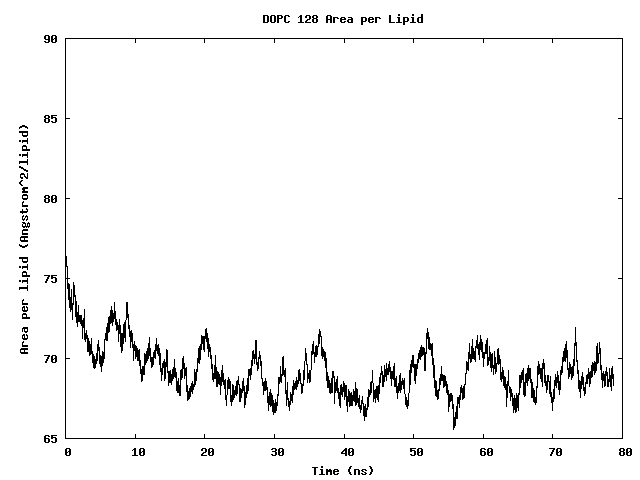
The area per lipid of the bilayer system is seen to converge after some simulation time.
Electron-Density Profile
The electron density profile provides a time-averaged measurement of the density of electrons through the lipid bilayer. This can be calculated throughout a trajectory in a fairly straightforward manner. Electron density plots are often compared with experimental models of the electron density.Lipid analysis functions have been added to the cpptraj program. cpptraj is now capable of calculating an electron-density profile and lipid order parameters.
In order to calculate the electron density with cpptraj, an script input file for cpptraj is needed.
A sample input file is provided below:
density.cpptraj
# NOTETo run the modified ptraj executable script, use:
# Your frames will be different depending on the part of
# the trajectory you choose to analyze
# Load the trajectory file frames 2500-7500 inclusive
trajin ../05_Prod.nc 2500 7500 1
# Center the lipid bilayer tail residues at zero coordinates
center ":PC | :OL" origin
# Image all water molecules to the zero coordinates using the
# center of mass of the molecules
image origin center ":WAT"
# Calculate the electron density using 0.25 Angstrom slices.
# Write out to the file electron_density.dat
density out electron_density.dat electron delta 0.25 "*"
ptraj_mod DOPC_128.prmtop < density.ptraj_modNow you have a file electron_density.dat with the electron density.
Plot with your favorite program. For example, in the gnuplot command line:
$ gnuplot
> plot "electron_density_normalized.dat" using 1:2 with lines
Electron Density Profile for 128 DOPC Bilayer
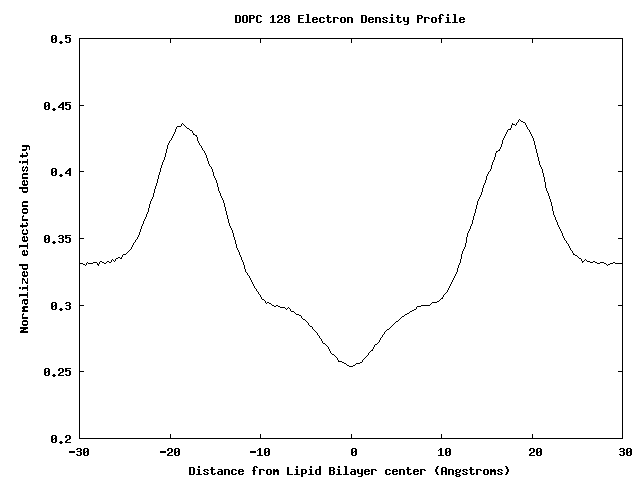
Membrane-Bound Proteins
Rhodopsin Embedded in a 2:2:1 POPC:POPE:Cholesterol Lipid Bilayer
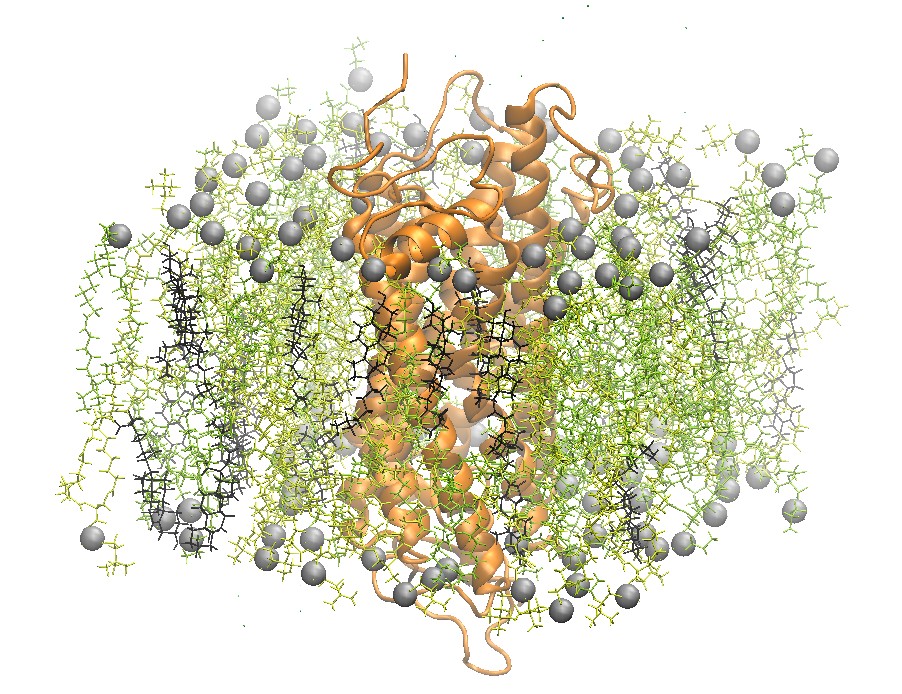
One recently studied membrane-bound protein system is that of rhodopsin, the visual pigment in photoreceptor cells. Rhodopsin was one of the first G-Coupled Protein Receptors (GPCR) to be crystallized. Grossfield et al. used Blue Gene, one of the top supercomputers at the time, to study rhodopsin on extremely long time scales. In particular, the authors were able to examine the dynamics of internal water molecules in the receptor and their effect on the function of rhodopsin [5].
In this section, a summary of the process to build a starting structure and prepare a molecular dynamics simulation for rhodopsin is outlined.
Rhodopsin (1U19)
Rhodopsin (1U19) Crystal Structure with Water Molecule Oxygens and Retinal Shown
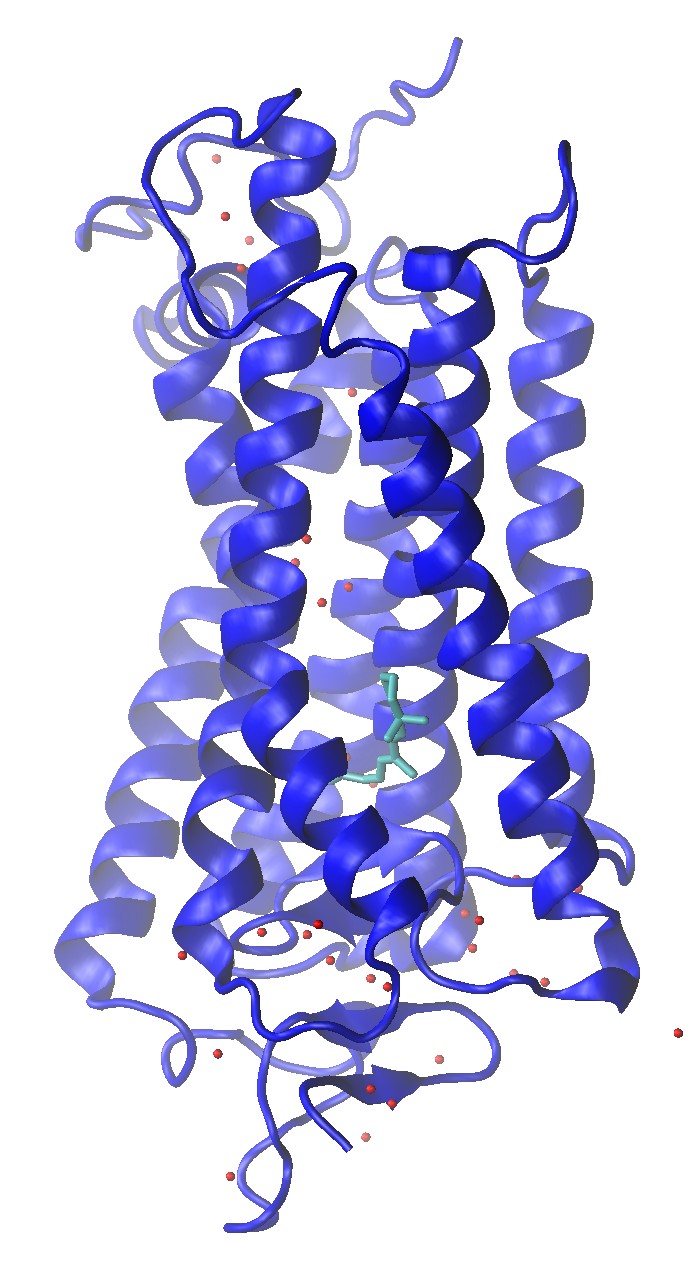
Build a Rhodopsin 2:2:1 POPC:POPE:Cholesterol System
The crystal structure for rhodopsin is available at the RCSB PDB database, ID 1U19.For molecular dynamics of this system, it is possible to again use the CHARMM-GUI web site to build the structure.
Select Protein/Membrane System for your membrane-bound protein and enter the RCSB PDB ID of rhodopsin and choose RCSB from the structure type.
Model/Chain Selection Options
You'll need to choose the chain from the structure file. In this case, choose just the chain A for the initial structure.
Continue to the next step.
Note
In a more detailed simulation, the retinal molecule should be included in the simulation. This tutorial will not cover how to prepare parameters for retinal.
PDB Manipulation Options
In the next step, the PDB must be further processed.
The ACE residue may be renamed to nothing to remove it from the structure. There should be a disulfide bond between cysteine residues 110 and 187.
Continue to the next step.
Computed Energy, Orientation Options, Positioning Options, Area Calculation Options
The rhodopsin structure must now be oriented in the Z direction for the lipid bilayer structure.
For the orientation of the protein, use the Align the First Principal Axis Along z.
Continue to the next step.
Calculated Cross Sectional Area, System Size Determination Options
This page now shows the cross sectional area of the protein within the bilayer. In the Grossfield paper, the authors use a mixture of 2:2:1 SDPC:SDPE:Cholesterol.
Because SD is not supported by Lipid14 currently, choose 50:50:25 POPC:POPE:Cholesterol.
Continue to the next step.
Determined System Size, System Building Options, Component Building Options
In the Grossfield paper, the authors used an ionic concentration of 14 sodium ions and 16 potassium ions for their system.
Set the ionic concentration to 0.07 M KCl for this simulation.
Continue with CHARMM-GUI with the default settings until the completion of step 5. After this, the system is assembled and ready to be downloaded.
Side and Top View of Rhodopsin in 2:2:1 POPC:POPE:Cholesterol Lipid Bilayer
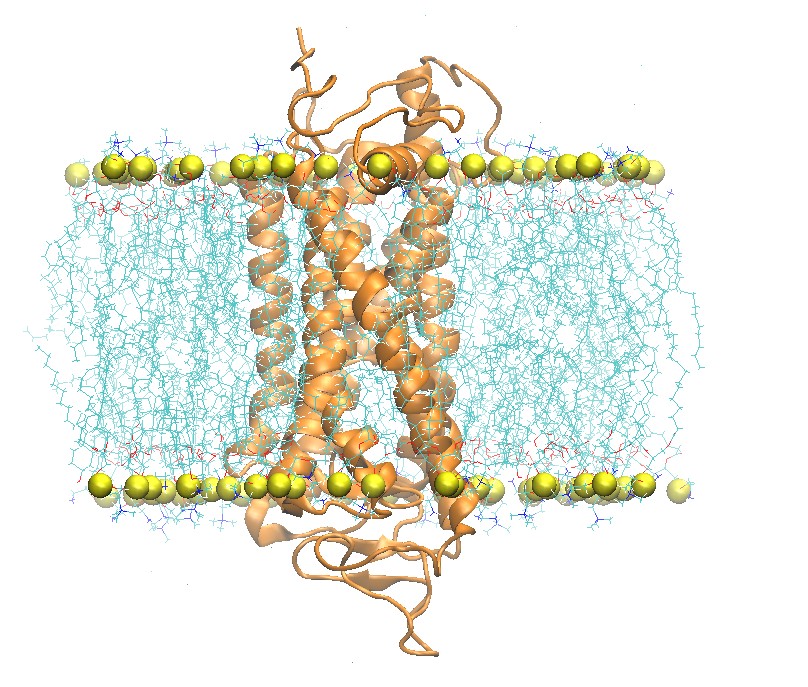
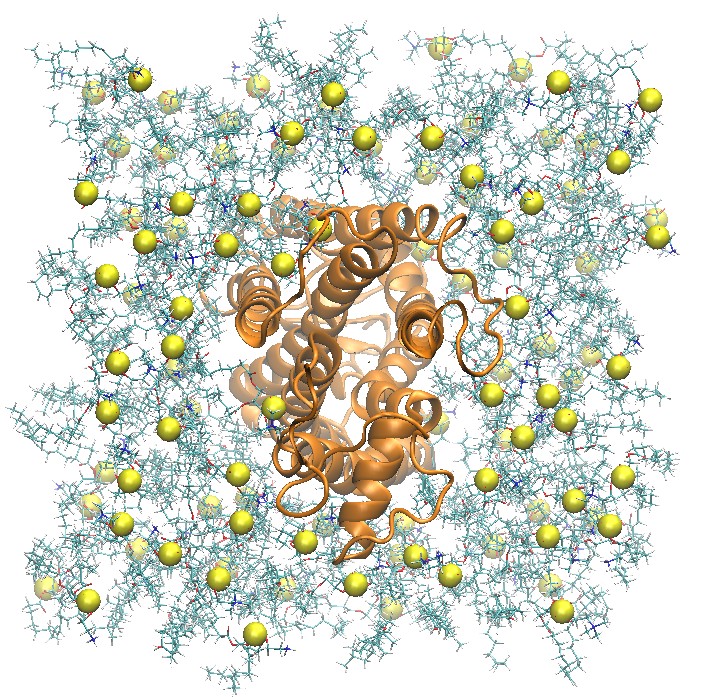
step5_assembly.pdb
This formatted PDB file can now be processed via the script charmmlipid2amber.py.
Warning
In this example, the CHARMM-GUI does not include a TER card after the protein chain. TER cards ending chains are part of the PDB format standard and must be added to the PDB file before processing with charmmlipid2amber.py.
Once the file has been processed, the next step is to start LEaP. In LEaP, a force field for the protein can be loaded , such as ff12SB along with the Lipid14 force field. Then it is possible to load the structure and build the parameter and topology file in LEaP.
Additional Resources
In addition to this tutorial, several other resources are available. Please refer to the following web sites for further information:- Amber Manuals
A reference for installing and using Amber software
- Amber Tutorials
Detailed step-by-step guides to specific tasks with Amber software
- Amber Mailing List
An active mailing list with archives since 1999
References
[1] Dickson, C.J., Madej, B.D., Skjevik, A.A., Betz, R.M., Teigen, K., Gould, I.R., Walker, R.C., "Lipid14: The Amber Lipid Force Field", J. Chem. Theory Comput., 2014, 10(2), 865-879.DOI: 10.1021/ct4010307
[2] Skjevik, A.A,; Madej. B.D.; Walker, R.C.; Teigen, K.; "LIPID11: A Modular Framework for Lipid Simulations Using Amber", Journal of Physical Chemistry B, 2012, 116 (36), pp 11124-11136.
DOI: 10.1021/jp3059992
[3] Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R., "GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid.", Soft Matter, 2012, 8, 9617-9627.
DOI: 10.1039/C2SM26007G
[4] Kucerka, N.; Tristram-Nagle, S; Nagle, J. J. Membrane Biol. 2005, 208, 193-202.
[5] Alan Grossfield, Michael C. Pitman, Scott E. Feller, Olivier Soubias, Klaus Gawrisch,
Internal Hydration Increases during Activation of the G-Protein-Coupled Receptor Rhodopsin,
Journal of Molecular Biology, Volume 381, Issue 2, 29 August 2008, Pages 478-486,
ISSN 0022-2836.