


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008
Building your own Custom Residues (old version) - SECTION 2
By Ross Walker
Stage 2 - Creating the non-standard CUA unit.
If simply loaded our edited pdb file above into XLeap at this point the vast majority of the file would load ok. There would be problems, however, with our non-standard CUA residue. Before we can successfully load our 1PLC_modified_final.pdb file into XLeap we need to tell it what our non-standard unit is.
We have several options on how to deal with this. The easiest option for a simple molecule would be to use Antechamber as you will do in tutorial 5. Antechamber provides an automated method for creating non-standard units. However, it only works with complete molecules, not fragments as we have here.
A second option would be to simply edit the residues within xleap this once and then disregard them when we quit. For this simple case of just the copper atom this is the quickest method but doesn't allow for any re-use of our residues. E.g if we wanted to create a second very similar protein we would have to go through and repeat the process of editing the residues in xleap all over again. While this would not be a problem with just the copper it is probably best if we learn how to do it in a way that is transferable to more complex systems.
The third option, and the one we will use here, is to create a new library file for the non-standard unit. In this way we can re-use the unit, by simply loading the library files into xleap before we load our protein pdb, for similar proteins. For big co-enzymes, such as NADH, this is the recommended method.
So, if you haven't already, fire up xleap and load our 1PLC_mod_final.pdb file into a new unit called PLC.
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
> 1PLC=loadpdb 1PLC_mod_final.pdb
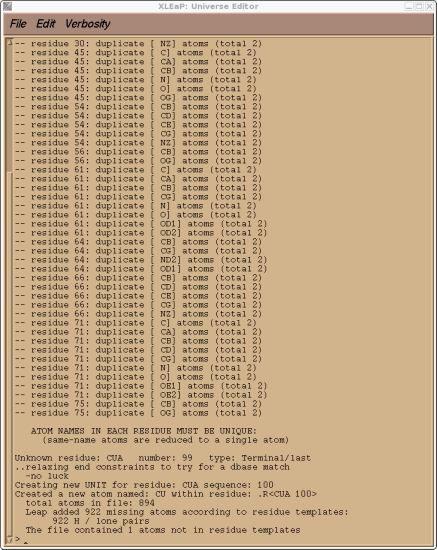
We should check the messages xleap gives us to ensure it has done what we expected. It should report only one unknown residue, our CUA. It should also have added any missing protons. Hence we expect xleap to add a total of 922 H atoms. Verify that this is indeed the case. If it isn't then you missed something during the pdb edit. There will also be a number of warnings about duplicate atom names, this is because we have have several residues with alternate conformations. These are just warnings, we don't need to worry about them, Leap will have just used the first conformation. This is just a test to check everything is okay. We can quit xleap now.
Creating CUA unit
We now need to create a library file for our CUA unit. The quickest way to do this, since we need an initial structure for this unit, is to simply cut it out of the 1PLC_mod_final.pdb file and save it as its own pdb file. This might seem crazy for copper and indeed it probably is but by doing it this way you learn a method that is transferable to much complex systems.
Now we can go ahead and load xleap again (are you getting good at this yet? :-)) and load the pdb file into its own unit:
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
> CUA = loadpdb cua.pdb
Now we need to tell xleap about our non-standard residue. If we had several atoms we would need to tell it which atom was bonded to which, we could either do this by hand in the edit window or use the bondByDistance command in Leap. However, since our copper unit contains only one atom we can skip this step.
> edit CUA
This should bring up our CUA residue in the editing window, you can turn on atom names with the Display menu.
The next step is to specify the atom types and charges.
Typically you would need to calculate atomic charges for the copper atom and all of the other atoms in our attached residues. This is necessary because the presence of the copper atom will alter the electron distribution in these surrounding residues. For AMBER this is done using the Restrained Electrostatic Potential Method (RESP). Details of calculating RESP charges can be found on the AMBER website. I will not cover this step in this tutorial. Instead we will just assume that the copper atom has a +1 charge and does not affect the neighbouring units. Note, the RESP fitting procedure has now been automated with a free program called R.E.D. Details of how to obtain and use R.E.D are available at http://www.u-picardie.fr/labo/lbpd/RED/.
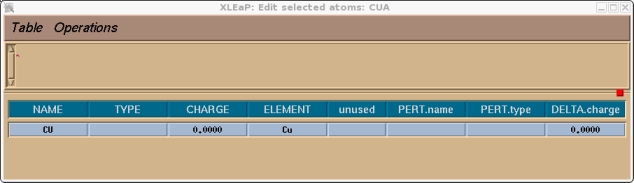
For the purposes of this tutorial we will use the charges shown in the screen shot below. In order to specify the charges and atom types we need to select the entire unit. Hit the select button in the Manipulation bar and then rubberband the entire molecule. It should all change colour. Then go to the Edit menu:
Edit->Edit Selected Atoms
The following box should come up.
You should now go through and assign an atom type and charge for all of the atoms in the unit, in this case just the copper atom. We will select CU for the copper atom type, this is not currently in use.
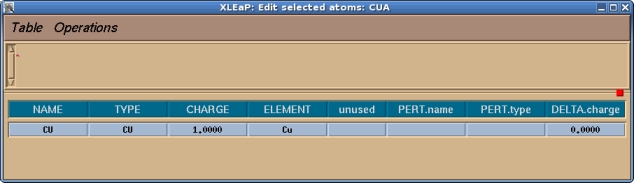
Once this is done we can select Table->Save and Quit. We can then close the edit window we should just be left with the command window. Typically the last thing we would need to do before we can save a library file for our completed unit would be to tell xleap what is the head atom for this unit and what is the tail atom. This information is used in order to connect the protein back bone. E.g. if we type desc MET we will see that there is a head and a tail defined:
> desc MET
UNIT name:
Head atom: .R<MET 1>.A<N 1>
Tail atom: .R<MET 1>.A<C 16>
.
.
.
However, if we look at our CUA we see that there is no head or tail atom defined.
> desc CUA
UNIT name: CUA
Head atom: null
Tail atom: null
.
.
.
Since we will not be making our copper residue part of the protein chain this is fine, however, if you were creating a residue that would form part of the protein chain you would need to use the set command in Leap to indicate which atom is the head atom and which the tail atom in the new unit.
We can now save the completed library file:
> saveoff CUA cua.lib
Here it is: cua.lib
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008