


Copyright (C) Pengfei Li & Kenneth M. Merz Jr. 2015
ZAFF Modeling Tutorial
Zinc AMBER force field (ZAFF) was developed by Peters et al.[1] in 2011. It was designed for 4-coordinated zinc metal centers (not suitable for 5- or 6- coordinated zinc centers). Users can use it directly for a system which has a similar coordination mode with these metal centers. In terms of the parameter files, the ZAFF.prep file is the prep file (which contains the residue name, atom name, atom type and charge information, please download it from this webpage for your modeling) and ZAFF.frcmod is the parameter file (which contains the bond, angle, torsion and VDW parameters, please download it from this webpage for your modeling) for the modeling. These parameters were generated using the Seminario/ChgModB method. These files can be used in tleap for straightforwardly build models. Here is a quick tutorial about ZAFF.
The following table lists the metal centers that had been parameterized in ZAFF.
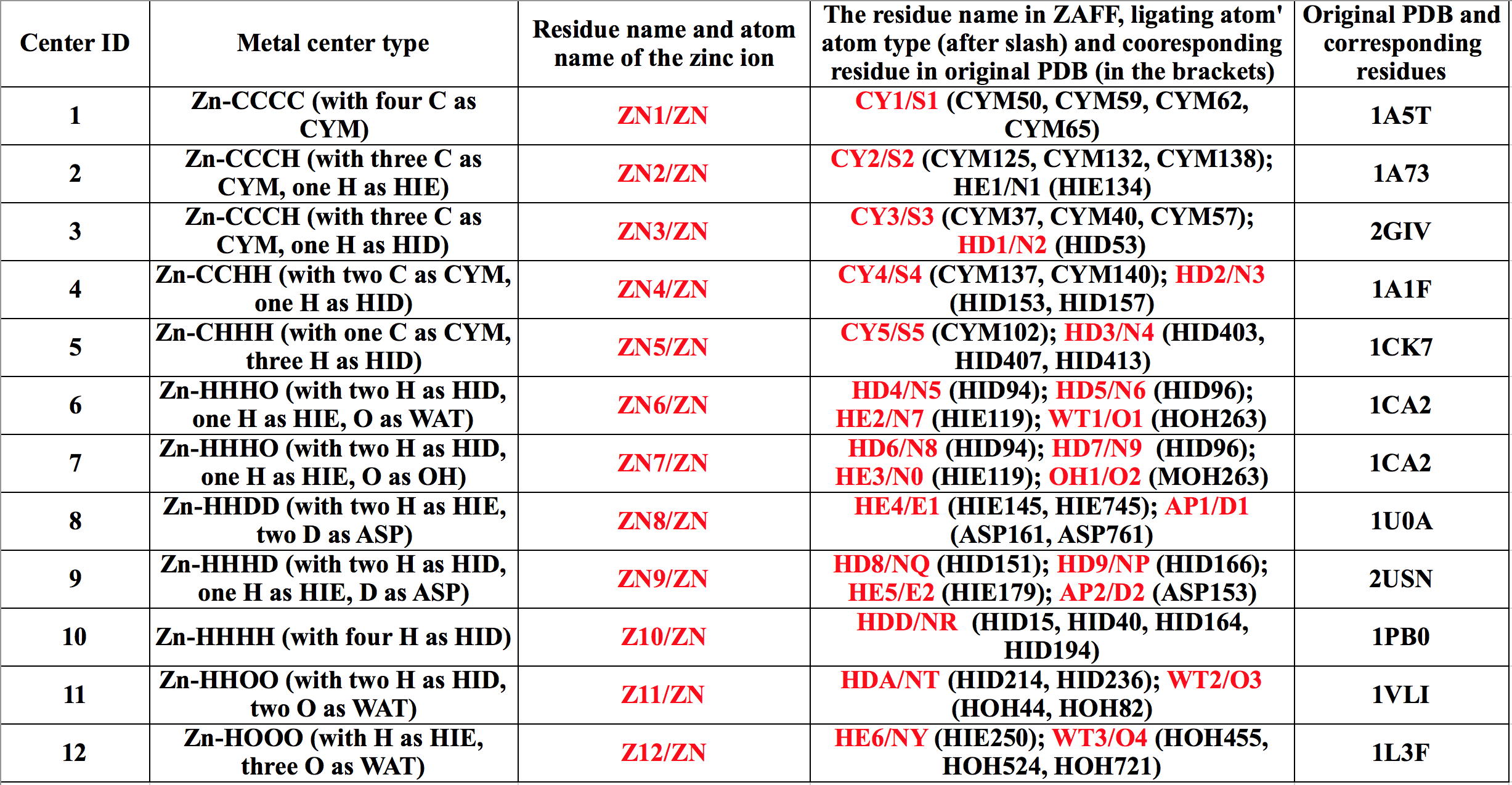
In ZAFF, all the zinc ions have atom type "ZN" while their VDW parameters are from Merz.[2] The VDW parameters of the zinc ion for PME simulation have been developed by Li et al.[3] in recent years. Users can modify the ZAFF.frcmod by themselves if want to use the new VDW parameters. Before doing the modeling, users need to check which metal center they want to simulate and then answer the following two questions:
1. Does the metal center you want to simulate have two water molecules bound to the same metal ion?
If yes, please use the nonbonded model to model the ion. Using the bonded model to simulate such system may cause distorted structures. This is because the torsion terms inside the metal site in ZAFF were treated as zero. Meanwhile, the water hydrogen was usually treated with a zero VDW interaction with surrounding particles so there will be no VDW repulsion when the oxygen and hydrogen atoms in different water molecules become very close with each other. This will cause a strong attraction between the hydrogen atoms on one water and the oxygen on the other water that bonds to the same metal ion (hydrogen has a positive charge while oxygen has a negative one), which can cause a distorted structure during the simulation. This was also proposed by Hu and Ryde.[4] Therefore, the metal centers with Center ID 11 or 12 in ZAFF are not suggested to use directly. Users can read this tutorial first and apply an restrainted nonbonded model afterwards (here is the restrainted nonbonded model tutorial).
2. Is the metal center I want to simulate included in the ZAFF?
If yes, answer the third question. If no, go to the MCPB or MCPB.py tutorial webpage for metal ion modeling from scratch.
3. What is the closet metal center in ZAFF to the metal center I want to simulate?
There are two reminders before answering this question.
(1) First, there may be two metal centers that have have the same metal center type but different protonation states. For example, there are two Zn-CCCH centers (with Center ID's of 2 and 3) where for the first one the H represents a HIE residue while for the second H indicates a HID residue. Users need to determine the protonation states of the binding residues first and decide which metal center should be used.
(2) Moreover, subtle structural characteristics may be another issue that needs to be considered to determine which metal center needs to be used. For instance, for the clusters with Center ID's of 6, 7, 9 there are two HID residues which are treated differently during the parameterization. From the Table above we can see that HD4 is the first HID residue (HID91) in the metal site. By checking 1CA2.pdb we can see the backbone of HID91 has hydrogen bonds with the only HIE residue in the metal site (HIE119). So for a system which has two HID residues and one HIE residue in the metal site, the HID which forms hydrogen bond with HIE residue should be considered as HD4 while the other should be considered as HD5.
Here we treat PDB entry 2GIV as an example to show how to perform the modeling with ZAFF in tleap. It is a histone acetyltransferase 1, which has a Zn-CCCH (H as HID) cluster (with Center ID as 3) in the metal site.
1. Clean up the PDB and adding hydrogen atoms
First we can download the PDB file 2GIV.pdb and then clean it.
awk '$1=="ATOM" || $1=="HETATM" || $1=="TER" || $1=="END"' 2GIV.pdb > 2GIV_clean1.pdb
Here we delete some residues in the PDB file to facilitate the modeling (here we simply show how to do modeling with ZAFF while in actual research these residues may need to be kept and extra modeling steps using antechamber may be needed). Here we delete the residues between ALY101 to PRO274 (terminal standard residue) and the ligand ACO in the PDB file because ALY is a non-standard residue while ACO is a ligand. We will keep the CL501 (which is Cl- ion) due and the HOH residues (which are waters) due to AMBER has atomic ions' libary and solvents' library which can recognize them respectively. Here is the PDB file after deleting these residues: 2GIV_clean2.pdb.
Then we can use pdb4amber to renumber the residue IDs and atom IDs in the PDB file:
pdb4amber -i 2GIV_clean2.pdb > 2GIV_clean3.pdb
During the modeling in actual research, users need to determine the protonation state of HIS because there are three histidine groups with different protonation states in the AMBER force field: HID, HIE and HIP. If you keep HIS residue name in the PDB file and perform the modeling using tleap, it will assign all the HIS residues as HIE by default. Users can use the H++ webserver to determine the protonation state of the HIS residues and add hydrogens to the PDB file before performing the following modeling. Users can check an example in the MCPB.py tutorial for details.
2. Check the structure and modify the PDB to the ZAFF format:
When opening the 2GIV_clean3.pdb file (the residue and atom IDs are renumbered by pdb4amber) using visualization software (such as VMD, Chimera, xleap etc.), we can see that there are four residues coordinated with the Zn ion. These residues are CYS34, CYS37, HIS50 and CYS54 while the zinc ion is ZN98. We need to modify these residue names to the residue name ZAFF uses. Please check the first table in this webpage. We can see that 2GIV has three CYS groups that all have residue name "CY3", a HIS residue group with a residue name of "HD1" and a zinc ion with residue name "ZN3". Then we change the residue names of residues 34, 37 and 54 from "CYS" to "CY3", change the residue name of residue 50 from "HIS" to "HD1", and change the residue name of residue 90 from "ZN" to "ZN3" in the PDB file. Please make sure there is a "TER" card between the protein and the Zinc ion. If not, tleap will include a bond between the protein and the zinc ion, causing trouble for the subsequent modeling. Here is the final PDB after these modifications: 2GIV_ZAFF.pdb.
3. Perform the modeling using tleap:
Here we use tleap to perform the modeling. We use the most recent AMBER force field ff14SB to build the model.
Here is the explanation of the tleap input file:
source leaprc.protein.ff14SB #Source the ff14SB force field for protein
source leaprc.water.tip3p #Source the TIP3P water model for solvent
addAtomTypes { { "ZN" "Zn" "sp3" } { "S3" "S" "sp3" } { "N2" "N" "sp3" } } #Add atom types for the ZAFF metal center with Center ID 4
loadamberparams frcmod.ions1lm_126_tip3p #Load the Li/Merz 12-6 parameter set for monovalent ions
loadamberprep ZAFF.prep #Load ZAFF prep file
loadamberparams ZAFF.frcmod #Load ZAFF frcmod file
mol = loadpdb 2GIV_ZAFF.pdb #Load the PDB file
bond mol.98.ZN mol.34.SG #Bond zinc ion with SG atom of residue CYM34
bond mol.98.ZN mol.37.SG #Bond zinc ion with SG atom of residue CYM37
bond mol.98.ZN mol.50.NE2 #Bond zinc ion with SG atom of residue HID50
bond mol.98.ZN mol.54.SG #Bond zinc ion with SG atom of residue CYM54
savepdb mol 2GIV_ZAFF_dry.pdb #Save the pdb file
saveamberparm mol 2GIV_ZAFF_dry.prmtop 2GIV_ZAFF_dry.inpcrd #Save the topology and coordiante files
solvatebox mol TIP3PBOX 10.0 #Solvate the system using TIP3P water box
addions mol CL 0 #Neutralize the system using Cl- ions
savepdb mol 2GIV_ZAFF_solv.pdb #Save the pdb file
saveamberparm mol 2GIV_ZAFF_solv.prmtop 2GIV_ZAFF_solv.inpcrd #Save the topology and coordiante files
quit #Quit tleap
In the above input file, the atomic_ions.lib file in $AMBERHOME/dat/leap/lib/ directory will be loaded when source the ff14SB force field (user can check the $AMBERHOME/dat/leap/cmd/leaprc.ff14SB file to see which lib files are loaded for the ff14SB force field). There are CL unit inside the atomic_ions.lib file therefore we can perform "addions mol CL 0" command in the input file to neutralize the system with Cl- ions. If the total charge of the protein system is negative, we can use "addions mol NA 0" command in the input file to neutralize the system with Na+ ions while the NA unit is also contained in the atomic_ions.lib file. If you create a lib file for metal ion(s) by yourself, please use a different name from the "atomic_ions.lib" file, preventing conflict with the existing file. Here we can perform the command:
tleap -s -f 2GIV_ZAFF_tleap.in > 2GIV_ZAFF_tleap.out
Finally we can use the generated topology file (2GIV_ZAFF_solv.prmtop) and coordinate file (2GIV_ZAFF_solv.inpcrd) to carry out the minimization and MD simulations.
References:
[1] Martin B. Peters, Yue Yang, Bing Wang, László Füsti-Molnár, Michael N. Weaver, and Kenneth M. Merz, Jr. "Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF)", J. Chem. Theory Comput., 2010, 6, pp. 2935-2947
[2] Kenneth M. Merz, Jr. "Carbon Dioxide Binding to Human Carbonic Anhydrase II", J. Am. Chem. Soc., 1991, 113, pp. 406-411
[3] Pengfei Li, Benjamin P. Roberts, Dhruva K. Chakravorty, and Kenneth M. Merz, Jr. "Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent", J. Chem. Theory Comput., 2013, 9, pp. 2733-2748
[4] LiHong Hu and Ulf Ryde "Comparison of Methods to Obtain Force-Field Parameters for Metal Sites", J. Chem. Theory Comput., 2011, 7, pp. 2452-2463