


Copyright (C) Pengfei Li & Kenneth M. Merz Jr. 2015
Building Bonded Model for A Ligand Binding Metalloprotein with MCPB.py
Newest version
The newest version of MCPB.py has been released in AmberTools, which is free of charge. And there is a binary distribution that can be installed through miniconda. Details can be found at here. The newest version is suggested to be used since it has the most robust support for metal ion modeling.
Reference
To cite MCPB.py please use the following reference:
Pengfei Li and Kenneth M. Merz, Jr., "MCPB.py: A Python Based Metal Center Parameter Builder." J. Chem. Inf. Model., 2016, 56, 599-604 (One typo in the paper: should be step 2b (not 2c) is the default for step 2.)
Moreover, useful tips can be found in the following book chapter:
Usage
Usage of MCPB.py is described below.
Usage:
MCPB.py [flags] [options]
options: -i input_file -s/--step step_number
[--logf Gaussian/GAMESS-US output logfile]
[--fchk Gaussian fchk file]
-h/--help help message
Details of the usage could be found in the in the AMBER manual.
Question
A Frequently Asked Question (FAQ) page was added. Please check whether the list has your question(s). If you meet any questions about the modeling that is not in the FAQ list, you can send an email to the AMBER Mailing list (amber@ambermd.org, you need to subscribe the Mailing list first if you have not done this: AMBER Mailing list).
Example
MCPB.py can perform parameterizations for both the metalloprotein and organometallic compound systems. Besides the example shown below, there are another two examples shown in the supporting information of the MCPB.py reference paper, one is for metalloprotein and the other is for organometallic compound.
Meanwhile, there is another example of MCPB.py about parameterizing the HEME group:
Click here to go to: MCPB.py Example of a HEME metalloprotein
Here we use a Zinc enzyme system as an example (PDB ID: 1OKL), which has 3 His residues and 1 MNS (5-(DimethyLamino)-1-Naphthalenesulfonamide) ligand in its metal site. Please make a new directory and put all the process files into it. MCPB.py will need some intermediate files (e.g. fingerprint files) during the modeling. For the system which has the metal ion inside a residue (like Fe ion inside the HEME residue), user can extract the Fe into a independent residue to perform the modeling. Or bugs may be proposed during modeling due to the specific software framework.
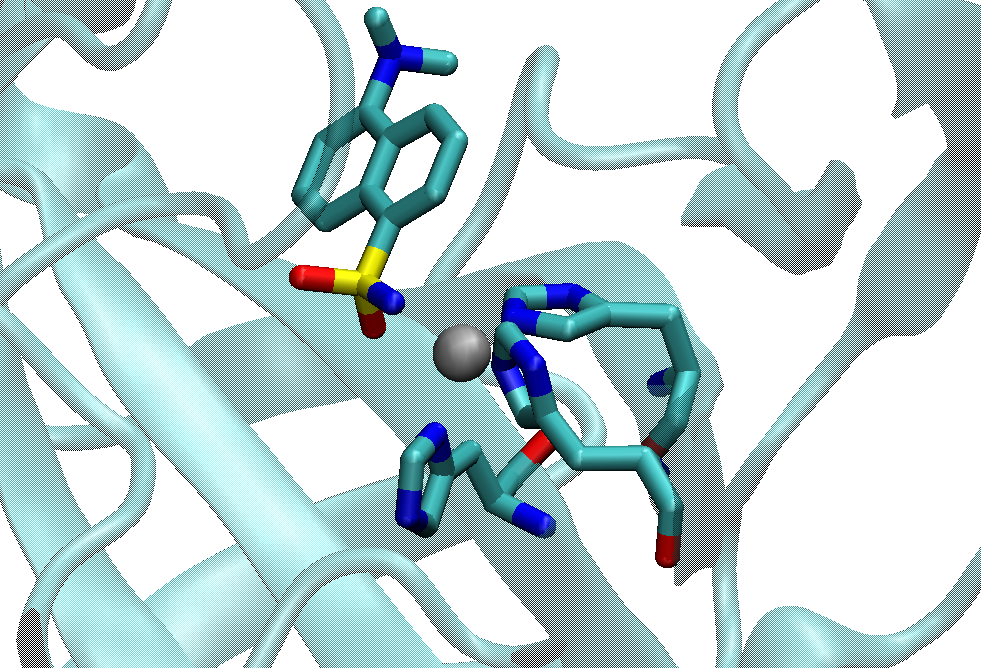
1. Prepare the PDB and mol2 files (effort: < 1 hour):
Generally we need a PDB file of the protein, which should employ the format of PDB version 3.0 and be consistent with the AMBER naming scheme for residues (e.g. HID, HIE, HIP for HIS). We also need to prepare mol2 files for non-standard residues (such as the metal ion, water and ligand).
Here is the original PDB file: 1OKL.pdb.
This step is SIGNIFICANT for a successful modeling, in which people are more likely to make mistakes than other steps, please read the following carefully.
For the PDB file, please make sure there are no atoms use the same atom name in a certain residue, which situation would be more likely to meet in a ligand residue. If there are, please manually correct it before processing the following procedures.
MCPB.py now supports more than 80 metal ions (see Figure 3 in the reference paper). For all of these metal ions and halide ions (if in the ion format but not neutral format), capitalized element name are suggested to use for both the residue name and atom name. Please make sure the residue name and atom name of the metal ion (not only for zinc used here) are all captilized (e.g. herein all equals "ZN", not "Zn", "zN", or "zn"), only in this way MCPB.py will recognize it as a metal ion. Meanwhile, please make sure each metal ion or halide ion (if it is in its ion format but not neutral format) is treated seperately as an indepedent residue in the PDB file. If you have metal site which has metal ion embeded in the ligand residue, for example, the HEME group in PDB file, please seperate the metal ion into an independent residue (also, with an unique residue number and atom number)in the PDB file. Meanwhile, in order to make MCPB.py recognize the atoms well, if you have atoms have atom names capitalized with numbers in your original PDB file (applies for protein or ligand), please change these atom names to atom names capitalized with their element symbols (for example, change "2HA1" to "HA12", change "1HAA" to "HAA1") in the PDB file before performing following steps.
1) First we prepare the PDB and mol2 files for the non-standard residues:
Note that it is important to get the total charge right for each of these mol2 files. Because MCPB.py will add these charges together to determine the total charge for the small and large models, and applies charge restraint of the total charge for the large model during the RESP charge fitting step.
For the ligandHere we use the reduce software in AmberTools to add hydrogens to the ligand, users can also the graphic software like GaussianView to add hydrogens manually if necessary (e.g. the protonation state is wrong by using reduce).
We copy the ligand MNS into one single PDB file: MNS.pdb
Afterwards we use reduce to add hydrogens (if you are using other software to add hydrogens, please make sure that you have all the atoms of the ligand have different names in the PDB file generated):
reduce MNS.pdb > MNS_H.pdb
Here we delete one of hydrogens, which connect to the ligating nitrogen atom of the zinc ion. Here is the PDB file after modification: MNS_fixed_H.pdb
Then we can use antechamber to generate mol2 files for non-standard residues, here we are using the AM1-BCC charge method to generate the charges where the ligand has a charge of -1. We treat the atoms using GAFF atom types.
antechamber -fi pdb -fo mol2 -i MNS_fixed_H.pdb -o MNS_pre.mol2 -c bcc -pf y -nc -1
After checking the MNS_pre.mol2 file we note that there is no "du" atom type in the MNS_pre.mol2 file, so we can rename it to MNS.mol2 file (Reminder: the mol2 file name of the non-standard residue (which is ligand for this example) should be the same (in letter and capitalization) as its residue name in PDB file and with suffix as ".mol2" for MCPB.py modeling).
Then we can perform the following command to obtain the frcmod file for the ligand:
parmchk2 -i MNS.mol2 -o MNS.frcmod -f mol2
Here we used parmchk2 because it has an improved performance over parmchk. Reminder: Please make sure there is one and only one blank line after each parameter section (BOND, ANGLE, DIHEDRAL, IMPROPER, NONBOND) in the frcmod file.The frcmod file will be used in the leap modeling as well.
For the metal ionFor the metal ion, we can copy the ZN ion into one single PDB file (ZN.pdb), and then use a script metalpdb2mol2.py to generate the mol2 file. One may need to modify the first line of this script to specify the path of the python program one wants to use. This script will be included in the AmberTools 2020 release, so users of AmberTools 2020 do not need to download it. We can run the following command:
metalpdb2mol2.py -i ZN.pdb -o ZN.mol2 -c 2
Here "-c 2" means the ion has a charge of 2.0. Here is the mol2 file generated: ZN.mol2. The oxidation state of metal ion can be assigned by using this option or modifying its charge information in the mol2 file afterwards.
For waterIf you want to keep crystal water(s) during the modeling, you can copy it into one single PDB file (e.g. WAT.pdb herein) and then use tleap to add the hydrogen atoms to the water molecule(s).
tleap -s -f wat_tleap.in > wat_tleap.out
You will get a WAT_H.pdb file, which has hydrogen atoms added to the crystal waters.
And you can use antechamber to generate the mol2 files for the water molecule(s). Here we treat the atoms using AMBER atom types.
antechamber -fi pdb -fo mol2 -i WAT_H.pdb -o WAT.mol2 -at amber -c bcc -pf y
Here we can see that antechamber assgined the atom types of H1 and H2 in WAT.mol2 as "HO", we can change them to "HW" atom type (the hydrogen atoms in water molecule) manually. Here is the mol2 file after modification WAT.mol2.
2) Then we prepare the PDB file which includes all the standard residues.
We can use the webserver H++ (here we use the default settings) to add hydrogen atoms to the PDB file. H++ will delete the non-standard residues during the modeling process. Along with generating the PDB file with hydrogen atoms, it will also generate AMBER topology and coordinate files for the system. Then we can use the topology and coordinate files to generate PDB file, since this PDB file will use an AMBER naming scheme for the residues (e.g. HID, HIE, HIP but not HIS for Histidine). Here we perform the command:ambpdb -p 0.15_80_10_pH6.5_1OKL.top -c 0.15_80_10_pH6.5_1OKL.crd > 1OKL_Hpp.pdb
H++ will not consider the metal ion while adding hydrogen atoms. We find that the metal site has a HIP residue in it, which is not physically meaningful. We can manually modify the HIP-92 to HID-92 (by deleting the HE2 atom and changing the residue name from HIP to HID in the PDB file). Here is the PDB file after modification: 1OKL_Hpp_fixed.pdb
3) Finally we can combine the PDB files into a single PDB file.
In order to be consistent with the usual sequence in a PDB file, we place the standard residues first then the metal ion and then the ligand and then water (if any).cat 1OKL_Hpp_fixed.pdb ZN.pdb MNS_fixed_H.pdb > 1OKL_H.pdb
Afterwards we can use pdb4amber to renumber the PDB file.
pdb4amber -i 1OKL_H.pdb -o 1OKL_fixed_H.pdb
Now we have the PDB file 1OKL_fixed_H.pdb and mol2 files MNS.mol2 and ZN.mol2 for the next stage of modeling.
2. Generate the PDB, Gaussian, GAMESS-US and fingerprint modeling files (effort: several minutes):
We need to create the input file manually. Here is the input file 1OKL.in. Users can check the pages 287-291 in the AMBER 2016 reference manual for the input parameters used by MCPB.py. One can also use Gaussian09 or GAMESS-US other than Gaussian03 to perform the QM calculations. When using Gaussian09, please add "software_version g09" as a line into the 1OKL.in file. For using the GAMESS-US program, please add "software_version gms" as a line into the 1OKL.in file. Here we set large_opt equals 1 (default 0, means not optimizing the large model) to optimize the hydrogen postions of the large model for RESP calculation, due to the hydrogen positions may not be stable for the ligand. Here we use the ff14SB force field (default) to perform the modeling. We perform the following command:
MCPB.py -i 1OKL.in -s 1
In this step, the PDB and fingerprint files of the small, standard and large models will be generated. The Gaussian input files of the small and large models will also be generated. The Gaussian input file for the large model will perform the optimization for the hydrogen atoms first to correct any poorly placed hydrogen atoms. The standard model fingerprint file (1OKL_standard.fingerprint) contains the atom type information of the atoms in the standard model (the 3rd column is the original atom type while the last column is the final assigned atom type). The atom type of the metal ion (name begins with M) and ligating atoms (name begins with Y/Z) are renamed automatically to differentiate them from the other atom types in the AMBER force field. You can treat step_number as 1n to generate the fingerprint file without renaming any atom types and then modify it manually (before doing that, please check the AMBER force field parm*.dat files to make sure your metal ion and ligating atoms do not have existing atom types). The fingerprint file also contains the linkage information between metal ion and surrounding atoms in the end of the file with beginning letter as "LINK". Users can also manually modify them for their needs (e.g. if there are any linkage changes after QM geometry optimization).
3. Perform the Gaussian/GAMESS-US calculations:
Gaussian03
Here we will perform the Gaussian calculations using the Gaussian input files obtained in previous step. You can change the parameters in the input file as you prefer (e.g., number of CPUs, memory usage, method, basis set, implicit solvation model, etc.). Make sure you got the multiplicity right for these Gaussian input files (for both the small and large models). Wrong multiplicity may cause error results, failure of convergence, or other issues. Herein we used the B3LYP/6-31G* level of theory to perform the calculations for both the small and large models because of its high performance cost ratio. It gives good results for the parameterization in this example, however, other level of theory could be used if users met failure. Meanwhile, users can use different level of theories to perform the calculations for the small and large models. But same level of theory should be used for geometry optimization and force constant calculation of the small model in order to make sure a local minima found. In order to make sure the parameterization is reasonable, we suggest users to check the structure of the small model after the optimization. This is because some coordination bonds to metal may break after the geometry optimization (not for this example), and in this situation, the final parameterization results can be problematic, a different level of theory or other strategy (e.g. performing a multiple step optimization) can be tried in order to get meaningful results. In the Gaussian calculation for large model, we only perform the optimization for hydrogen atoms to save computation tasks. This setting could be changed by modifying the "large_opt" variable in the MCPB.py input file. We recommend users do the calculations parallel since the jobs may take a while. Here are a link on running Gaussian03 job: Gaussian03 .
Perform the geometry optimization, and then force constant calculation, and then generate the fchk file for the small model:
g03 < 1OKL_small_opt.com > 1OKL_small_opt.log
g03 < 1OKL_small_fc.com > 1OKL_small_fc.log
formchk 1OKL_small_opt.chk 1OKL_small_opt.fchk
Perform the Merz-Kollman RESP charge calculation for the large model:
g03 < 1OKL_large_mk.com > 1OKL_large_mk.log
Gaussian09
How to run Gaussian09 can be seen here: Gaussian09
Perform the geometry optimization, and then force constant calculation, and then generate the fchk file for the small model:
g09 < 1OKL_small_opt.com > 1OKL_small_opt.log
g09 < 1OKL_small_fc.com > 1OKL_small_fc.log
formchk 1OKL_small_opt.chk > 1OKL_small_opt.fchk
Perform the Merz-Kollman RESP charge calculation for the large model:
g09 < 1OKL_large_mk.com > 1OKL_large_mk.log
There is a bug in Gaussian09 rev B.01 when doing the Merz-Kollman population analysis. Please following the "Gaussian 09 fix" section in the Bug Fix Webpage to solve the problem.
GAMESS-US
Except the Gaussian program, GAMESS-US can also be used for the calculation. How to run GAMESS-US could be through here: GAMESS-US Tutorials .
The input files for small model are 1OKL_small_opt.inp and 1OKL_small_fc.inp. First, optimizing the structure:
rungms 1OKL_small_opt 01 2 1 >& 1OKL_small_opt.log
After the job finished, manually copy the optimized coordinate from 1OKL_small_opt.log into 1OKL_small_fc.inp file and then perform the force constant calculation:
rungms 1OKL_small_fc 01 2 1 >& 1OKL_small_fc.log
The input file for the large model is 1OKL_larger_mk.inp. We can calculate the large model using following command:
rungms 1OKL_large_mk 01 2 1 >& 1OKL_large_mk.log
After the QM optimization of small model, if there is a coordination bond between metal and a ligating atom breaks, one may need to change the method or basis set to redo the calculations. If this problem can not be solved after several trials, one can still use the MCPB.py to facilitate the modeling but use another method (like PES scanning) to obtained metal involving bond and angle parameters and manually added it to the PREFIX_mcpbpy.frcmod file and carefully check the finally tleap input file to make sure everything is going well. If you parameterize a zinc metal site and met the above problem, you can use an empirial method to determine the metal involving bond and angle parameters (just treating step number as 2e instead of 2).
4. Perform the final modeling (effort: several minutes):
5. Check the modeling (IMPORTANT for a successful modeling):Herein we use the Seminario method to generate the force field parameters. Other options are available: Z-matrix (with step_number 2z) and Empirical (with step_number 2e) methods. The Empirical method doesn't need any Gaussian calculations to obtain the force constants (but still needs Gaussian calculation for getting the RESP charges), but it only supports zinc ion modeling in the current version.
MCPB.py -i 1OKL.in -s 2
We can get a 1OKL_mcpbpy.frcmod file, which will be used in the leap modeling.
Herein we use the ChgModB to perform the RESP charge fitting and generate the mol2 files for the metal site residues. Other options are also available: ChgModA, ChgModC and ChgModD (as 3a, 3c and 3d respectively).
MCPB.py -i 1OKL.in -s 3
In this step we can get 5 mol2 files for the metal site: HD1.mol2, HD2.mol2, HE1.mol2, ZN1.mol2 and MS1.mol2. The charges in these files are refitted by MK RESP charge fitting aglorithm. We will use these mol2 files in the leap modeling.
Generate the tleap input file:
MCPB.py -i 1OKL.in -s 4
Then we can obtain a new pdb file after renaming the metal site residue names (1OKL_mcpbpy.pdb) and a leap input file (1OKL_tleap.in). You can change the input file as you comfort with. Reminder: In this input file, the atomic_ions.lib file in $AMBERHOME/dat/leap/lib/ directory will be loaded when source the ff14SB force field (user can check the $AMBERHOME/dat/leap/cmd/leaprc.ff14SB file to see which lib files are loaded for the ff14SB force field). So we loaded the atomic_ions.lib twice in total, users can delete the "loadoff atomic_ions.lib" line in the input file, which would give same results. If you create a lib file for metal ion(s) by yourself, please use a different name from the "atomic_ions.lib" file, preventing conflict with the existing file. Afterwards we can use tleap to generate the topology and coordinate files:
tleap -s -f 1OKL_tleap.in > 1OKL_tleap.out
Now we have topology and coordinate files ( 1OKL_solv.prmtop and 1OKL_solv.inpcrd) generated.
Before performing the minimization and MD simualtions, we can use VMD and cpptraj to do a quick check about the modeling.
1) Firstly we can use VMD to do a check about whether the coordiante bonds to metal ion exist in the topology file.
We can use the following command :
vmd -parm7 1OKL_solv.prmtop -rst7 1OKL_solv.inpcrd
and then select Graphics->Representations and then type "same residue as within 3 of resname ZN1" and after that click the "Apply" button in the right down corner.
We can select Graphics->Colors and then select Display in the Categories, Background in the Names, 8 white in the Colors.
Then we can adjust the position and point of view, afterwards we can see that all the four coordination bonds to zinc ion, which validates our modeling.
2) Secondly, we can use cpptraj to do a check about the atom numbering issue by using following command:
cpptraj -p 1OKL_solv.prmtop
If there is no error report, we can go ahead to do minimization and MD simulations (actually there is no error reported for this case, so we can go ahead to do the minimization and MD simualtions).
*IF* there is error reported like follows (for some other cases but not for this one):
Error: Atom XXX was assigned a lower molecule # than previous atom. This can
Error: happen if 1) bond information is incorrect or missing, or 2) if the
Error: atom numbering in molecules is not sequential. If topology did not
Error: originally contain bond info, 1) can potentially be fixed by
Error: increasing the bondsearch cutoff offset (currently 0.200). 2) can be
Error: fixed by either using the 'fixatomorder' command, or using
Error: the 'setMolecules' command in parmed.py.
Error: Could not determine molecule information for 1OKL_solv.prmtop.
Error: SetSolventInfo: No molecule information.
Error: Could not determine solvent information for 1OKL_solv.prmtop.
We can use cpptraj to fix the atom order:
(Here is the input file: fixatord.in)
cpptraj -p 1OKL_solv.prmtop -c 1OKL_solv.inpcrd -i fixatord.in > fixatord.out
Then we can try the following command to see whether the problem solved:
cpptraj -p fixed.1OKL_solv.prmtopIf there are no error like showed above, we can use the fixed.1OKL_solv.prmtop and fixed.1OKL_solv.inpcrd to do the modeling.
3) Finally, we can use ParmEd to check the metal site parameters:
Here is the input file: mcpbpy_parmed.in, in which we have ":ZN1" as the mask of the zinc ion. Additional information could be found in the Amber Masks section in the chapter of Atom and Residue Selections in the AMBER manual. Here we use ParmEd and above input file to check the parameters:
parmed -i mcpbpy_parmed.in -p 1OKL_solv.prmtop
Generally the metal ion related parameters are follow these creteria:
A) The bond force constants between an metal ion and its ligating atoms are less than 200 kcal/(mol*Angstrom^2), and the eqlibirum bond distances are less than 2.8 Angstrom;
B) The angle force constants related to the metal ion are usually less than 100 kcal/(mol*Rad^2) while the equlibirum angle values are bigger than 100 Degree;
C) All or most of the dihedral potential barriers are zero for metal involved dihedrals;
D) The RESP charge of the metal ion are less than its oxidation state, usually even less than +1;
E) The LJ Radius of one metal ion is usually bigger than 1.0 Angstrom.
We can see the output results of ParmEd are consistent with the creteria we listed above, so we can go ahead to use the topology and coordinate files to perform related modeling.
If you had a distorted metal site structure after minimization or molecular dynamics simulation or met errors during these procedures, which might imply the normal bonded model fails to describe the metal site under this situaiton. For example, for the case that there are two water molecules connected to the same metal ion, which would cause structure distortion because of the 1-4 VDW interaction between oxygen in one water and hydrogen in the other is zero while the 1-4 electrostatic interaction between them are nonzero and attractive. The strategy is to delete related bonds in the LEaP modeling, treat the bonds in a nonbonded way, while use harmonic restraints for these bonds in the minimization and molecular dynamics simulations. In this way the full nonbonded interactions between two water molecules would be considered to solve the problem. Users can treat all the coordination bonds in this way or treat some of them in the bonded way while the others in this way (a hybrid bonded/restrained nonbonded model). An related tutorial could be found here: restrainted nonbonded model tutorial).
There is another tutorial about using the above AMBER force field in CHARMM. In this way users can take advantage of the functions provided by the CHARMM software package. Interested users can check this link.