


TIPs and FAQs of MCPB.py
By Pengfei Li, Zhen Li, and Kenneth M. Merz Jr., 2024
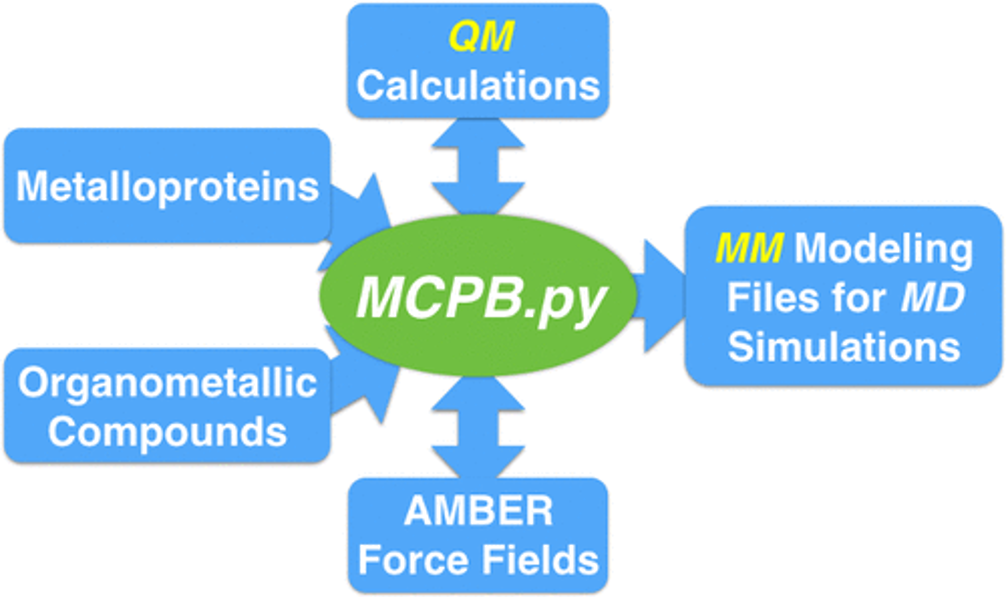
Figure 1.1: Overview of the MCPB.py workflow.
0. About preparation
Q0.1. What should I do if I cannot create a mol2 file for a metal ion using antechamber?
For example, you get the following Error Message:
antechamber: Fatal Error! Unrecognized case-sensitive atomic symbol (XX).
This is because antechamber could not recognize the atomic symbol for the metal ion, you can use the metalpdb2mol2.py program to do the conversion. You can check the “For the metal ion” section in this webpage for how to use the metalpdb2mol2.py program.
Q0.2. What should I do if I want to assign the metal ion to a specific oxidation state?
MCPB.py reads the charge of the ion from its mol2 file. If you want to assign the ion to a specific oxidation state, just manually modify the charge of the ion in the mol2 file would be fine, then you can go ahead for the MCPB.py modeling from the first step.Q0.3. About the PDB file, mol2 file(s) and their matching:
The following points are important to keep in mind. If you have the PDB file and mol2 file(s) in the correct format, it will save you a lot of time and frustration. Otherwise, you will waste a lot of time to debugging your MCPB.py modeling. You should have: (1) In your PDB file, it is critical to make sure each atom in the same residue has a unique atom name. In this way, MCPB.py will differentiate them by names. Otherwise, MCPB.py will handle these atoms incorrectly. This will negatively influence your simulation results. (2) Moreover, the atoms in your PDB file and mol2 file(s) should match each other. There should be a one-to-one match for these atoms (both residue name and atom name should exactly match in a case-sensitive manner) in the PDB and mol2 file(s). Otherwise MCPB.py will have issues assigning the atom types and charges to the atoms. A more detailed explanation about this can be found at here. We strongly suggest every user of MCPB.py to read this webpage. (3) Last but not the least, each mol2 file should have a file name (file name before the suffix) identical to the residue name it contains.
Q0.4. What if I want to deal with an unusual metal-ligand bond?
By default, MCPB.py will only recognize the metal-N/O/S/F/Cl/Br/I bonds but not metal-C/H bonds. This setting is to prevent assigning unphysical bonds based on low resolution structures. There is a variable “bonded_atom_pairs”, based on which you can set additional metal-ligand bonds manually (e.g. a metal-C bond). You can check the AMBER manual for details.
1. About Step 1
Q1.1. What should I do if I get the “KeyError” of the Atnum function?
For example:
> AtNum = Atnum[gatm.element] > KeyError: '1'
It means MCPB.py could not find the atomic number for the element based on the PDB file. MCPB.py determines the element of the atom based on the information in the PDB file. In particular, for the metal ions, it will determine the element based on the residue name and atom name. MCPB.py now supports more than 80 metal ions (see Figure 3 in the reference paper ). The detailed information of these metal ions are listed in the METAL_PDB dictionary in the $AMBERHOME/AmberTools/src/pymsmt/pymsmt/mol/element.py file. Three principles should be followed for the residue names and atom names in the PDB file:
(1) In general, for all of these metal ions and halide ions (if in the ion format but not neutral format), capitalized element names should be used for both the residue name and atom name. Please make sure the residue name and atom name of the metal ion (not only for zinc used here) are all capitalized (e.g. for zinc you should use "ZN", not "Zn", "zN", or "zn"), only in this way will MCPB.py recognize it as a metal ion.
(2) Further, please make sure each metal ion or halide ion is treated separately as an independent residue in the PDB file. If you have metal sites which has a metal ion embedded in the ligand residue, for example, the HEME group in the PDB file, please separate the metal ion into an independent residue (with a unique residue number and atom number) in the PDB file.
(3) Moreover, to make MCPB.py recognize the regular atoms (except the ions) in a non-standard residue you should change these atom names to start with their capitalized element symbols (for example, change "2HA1" to "HA12", change "1HAA" to "HAA1") before the performing following steps.
(4) For residue names and atom names in a standard amino acid residue, please follow the PDB format version 3. Further details are given below in the answer to Q1.4.
Q1.2. What should I do if I get the “KeyError” of the chargedict function?
For examples:
> chg = str(int(chargedict[i])) > KeyError: 'H2' OR: > chg = int(round(chargedict[ionname], 0)) > KeyError: 'FE' OR: >chg = int(round(chargedict[ionname],0)) >KeyError: 'C'*
This error concerns the KeyError of the chargedict dictionary. This is usually due to a mismatch between the PDB file and the mol2 file for an atom name, or it implies a mismatch between the ion_ids variable in the MCPB.py input file and the metal ion in the PDB file. In the above examples, the first example might mean MCPB.py could not find the atom with name H2 in the mol2 file. This atom appears in the PDB file but not in the mol2 file. The second example might indicate MCPB.py could not find the atom name FE in the mol2 file. There might be an inconsistency between the PDB file and mol2 file for the metal ion. Moreover, it also might be because the ion_ids variable setting in the MCPB.py input file was wrong, which should correspond to the metal ion instead of any other atom.
Q.1.3. What should I do if I met the “KeyError” of the mol.atoms[i] function?
For example:
> line 50, in get_ms_resnames > resid = mol.atoms[i].resid > KeyError: 7440
Such an error means MCPB.py could not find an atom with atom ID of 7440 in the PDB file, which is likely due to an incorrect ion_ids variable in the input file. ion_ids in the MCPB.py input file should correspond to the atom ID of the metal ion you are interested in.
Q1.4. What should I do if I met the “KeyError” of the libdict function?
For examples:
> attype = libdict[resname + '-' + atname][0] > KeyError: 'MET-AS' OR: > attype = libdict[resname + '-' +atname][0] > KeyError: 'ASP-HB1'
This indicates there is a mismatch between the PDB file and the AMBER force field library (for the standard residues) or mol2 file (for the non-standard residues and metal ions), so MCPB.py could not assign the atom type for a specific atom. (1) In the AMBER force field library, the residue names and atom names in the standard residues follow the PDB version 3.0 style, which means one should also use consistent settings in the PDB file used for the MCPB.py. We would suggest you use the H++ web server to add the hydrogen atoms, which will add the hydrogen with names consistent with PDB format version 3.0. If you are using another program (e.g. pymol) to add the hydrogen atoms, you may meet the above error. (2) The above error may be due to a mismatch between the PDB file and mol2 file, that it could not find the atom type information for a specific atom in the mol2 file. To solve this problem, one needs to make sure the PDB file and mol2 file are consistent with each other: for a specific residue, each atom in the PDB file should have a unique atom name, and the PDB file and mol2 file should have the residue name and atom name in an exactly matching manner for each atom. A more detailed explanation about such a matching can be found at here.
Q1.5. About Cl− ion recognition
For example:
> print('YES', atyp2 + massparms[atyp1], file=fmf)
> KeyError: ‘CL’
When handling a Cl− ion which is coordinated to the metal ion in the metal site, one needs to set its residue name as CL and atom name as CL in both the PDB file and mol2 file. And then the program will recognize it as a Cl− ion. Moreover, one needs to set the atom type as Cl (where L is in the lower case) and a charge of -1 in the mol2 file. This is because the ff19SB force field does not have the “CL” atom type, instead, it has the “Cl” atom type for chloride. Other halogen ions should be handled in a similar way.
Q1.6. About OW atom type recognition
For example:
> print('YES', atyp2 + massparms[atyp1], file=fmf)
> KeyError: 'OW'
This issue may be because the separation of the water model from the protein force field in the most recent versions of AmberTools, because of this MCPB.py could not find the water model parameters from the protein force field. The MCPB.py program in AmberTools24 will have this issue fixed. When you are using AmberTools23 or previous versions, to solve this problem (for illustrative purposes we are assume you are using the opc water model, if you are using another water model, you can adjust accordingly), you can copy the $AMBERHOME/dat/leap/parm/frcmod.opc file into your working folder, and then add "frcmod_files frcmod.opc” into your MCPB.py input file and do the MCPB.py procedure from the beginning.
Q1.7. mol2 file not provided:
For example
> pymsmt.exp.pymsmtError: LIG is required in naa_mol2files but not provided.
This issue is due to residue LIG being included in the PDB file, but its mol2 file is not provided after the naa_mol2files variable, so MCPB.py cannot assign atom types and charges to this residue. Moreover, the mol2 file should have its file name (file name before the suffix) the same as the residue name it contains. For the above example, its name should be LIG.mol2.
2. About Step 2
Q2.1. No Cartesian force constants found
For example:
> pymsmtexp.pymsmtError: There is no 'Cartesian Force Constants' found in the fchk file. Please check whether the Gaussian jobs are finished normally, and whether you are using the correct fchk file.
This is because the fchk file you generated does not have the Cartesian force constants information. The Cartesian force constants will be generated when you do a frequency analysis. However, it will not be generated when you optimize the structure only. To solve the problem, one should go back to check the Gaussian input file you lastly used to generate the chk file (based on which you converted to the fchk file), to see whether it performs a frequency analysis or not. If not, one needs to fix the issue to make it work.
Q2.2. Negative force constant(s) in the generated frcmod file.
This error may be due to different reasons.
(1) You did not get the Cartesian Hessian matrix based on an optimized structure. If the structure is not a local minimum, it could give you negative force constants for some bonds and angles.
(2) You have manually changed the atom sequence in the .com file before performing Gaussian calculations. Now it is not consistent with the atom sequence in the fingerprint file generated. To solve the problem, you should not manually change the atom sequence in the .com file when doing the Gaussian calculations.
(3) If you are sure the error is not due to the first two reasons, it is probably due to the limitation of the Seminario method. For such a case, you may try the Z-matrix method or the empirical method to generate the force constants.
Q2.3. What should I do if I met the key error of the attypdict function?
For example:
> attyp = attypdict[i][1] > KeyError: 11026
This error means MCPB.py could not find the atom type of an atom. This might due to that the mismatch between the PDB file and mol2 file, that MCPB.py can find the atom with this specific atom ID (i.e. 11026) but could not identify the atom type of it. Note that the residue names and atom names in your PDB file should match with those in the mol2 file(s). There should be a one-to-one match for these atoms (both residue name and atom name should be exactly match in the case-sensitive manner) in the PDB and mol2 file(s). Otherwise MCPB.py will have issue to assign the atom types and charges to the atoms. A more detailed explanation about such a matching can be found at here, I strongly suggest every user of MCPB.py to read this webpage.
3. About Step 3
Q3.1. IOError about the resp2.chg file
For example,
> IOError: [Errno 2]no such file or directory: 'resp2.chg'
This error says it could not find the resp2.chg file. The third step of MCPB.py performs RESP charge fitting. If it could not generate the resp1.chg file or resp2.chg file, it means it met an issue when performing the RESP fitting. This issue may be caused by that you used the small model to do the Merz-Kollman analysis instead of using the large model, which failed the RESP charge fitting due to such a mismatch.
Q3.2. The generated mol2 files have charges with wired values (e.g. huge magnitudes)
This might because the total charge was wrong in the Merz-Kollman analysis of the large model. MCPB.py will calculate the total charge of the system by adding up the charges of all the residues in the metal site (based on the ion and non-standard residue mol2 files you provide and the amino acids in the AMBER force field database), if this number is different from the real charge of your metal site, it will cause big error in the RESP charge fitting. I suggest you to check the charges and multiplicities you use for your small model and large model Gaussian calculations as well as the charge of the metal site MCPB.py assumes (which is obtained by adding up the charges of all the residues in the metal site, as discussed above). If there was anything wrong or inconsistent, you should fix the issue(s) and rerun the MCPB.py modeling from the first step (i.e. option -s 1) and also any erroneous Gaussian calculation (if the charge or multiplicity was wrong in the Gaussian calculation).
The above error might also because you have changed the sequence of *large_mk.com file, making its atom sequence mismatched with the fingerprint file generated. Such an inconsistency will cause wired RESP charges generated. If there was anything wrong or inconsistent, you should fix the issue(s) and rerun the MCPB.py modeling from the first step (i.e. option -s 1) and also any erroneous Gaussian calculation (if the charge or multiplicity was wrong in the Gaussian calculation).
Q3.3. Mismatch between the number of atoms and fitted charges
For examples:
> "Error: The length of coordinates and ESP charges are different!" OR: > “Error: the charges and atom numbers are mismatch for the standard model!”
Such a kind of error may be because you added/deleted atoms in the Merz-Kollman analysis of the large model. This caused inconsistency of the number of atoms in the coordinates and ESP charges. You should fix the issue(s) and rerun the MCPB.py modeling for this step (including the relevant Gaussian calculations). Such a kind of error may also be because one used the Gaussian09 rev B.01 for the Merz-Kollman population analysis, which generated output file having a different format from those generated by other Gaussian versions. If so, please check the "Gaussian 09 fix" term in this webpage for how to solve the issue.
4. About Step 4
Q4.1. Can I change the water model in the generated tleap input file?
The ion parameters usually depend on the water model. If one wants to use a water model different from the default one, it is better to change the water_model variable in the MCPB.py input file in the very beginning.
5. Other questions:
Q5.1. Can MCPB.py support simulations in GROMACS?
MCPB.py also supports simulations in GROMACS (with the help of ParmEd). For doing that, please check this book chapter.