


Implementing modified 12-6-4 LJ potential using ParmEd
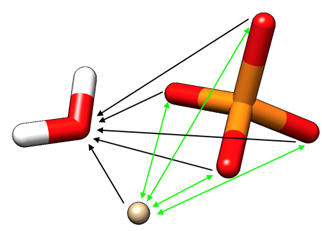
Figure 1.1: A phosphate-water-ion system with solvent C4 (indicated black) and modified C4 (indicated green) terms
Learning Outcomes
Overview
This is a tutorial to run an MD simulation using PMEMD.cuda in AMBER24 where metal->water, ligand->water, and metal<->ligand C4 interactions are applied. The model is called modified 12-6-4 (m12-6-4) because it does not have unwanted background C4s, and the wanted C4 interactions are all fine-tuned by parametrizing the polarizability of the ligands. Please go through this tutorial carefully to guarantee that the calculated energy be correct.
Introduction
In AMBER24's PRMTOP file, C4 interactions are described as non-directional. However, in PMEMD.cuda engine, C4 interactions are actually considered directional (see Figure 1.1). The engine will check whether an atom type has "+" or "-" in it (criteria #1) and whether it has a non-zero C4 to itself (criteria #2). If both are fulfilled, this atom type will be a C4 donor.
(For AMBER20 users, since the code has a built-in issue, criteria #2 is defined slightly differently: if the atom type has an index of "k" and a non-zero C4 to atom type with index "k+1", it will fulfill criteria #2. Note that "k" could be the last index for the atom type list. In this case, it will cause segfault because "k+1" is out of index in the atom type list.)
Then, the engine will apply C4 interactions onto all atom pairs with at least one end matching the donor atom type. In Figure 1.1, such C4 interactions are depicted as black arrows pointing from atoms with the donor atom type to other atoms within the VDW cutoff. For this system only, a final working C4 matrix will be as what Figure 1.2 shows.
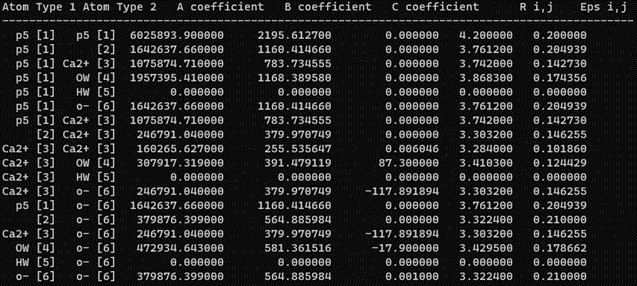
Figure 1.2: Final C4 matrix displayed in a pairwise style
Note here two things:
- It is possible to have C4s between two donor atom types. These are marked as green double arrows and double-calculated in the engine. To resolve this issue, the C4 value between two donors must be manually halved from the PRMTOP file (criteria #3).
- There is no one-direction C4 between Ca2+ and Phosphorus, and also no bi-direction C4 between phosphate oxygens. This is because that (Ca2+)->(P) C4 values are set to be zero by tuning the polarizability of Phospherus to be zero in the polarizability file pol_addc4_phosphate.dat, and (o-)<->(o-) values are manually set to be infinitely small (0.001).
Methods:
Step-by-step explanation:
- In "addc4_PO4.in", notice that we changed the "OP" atom type to add a new atom type for "o-". This is in order to fulfill criteria #1.
- In "pol_addc4_phosphate.dat", note the polarizability of Ca2+ is set to 0.001. This is to fulfill criteria #2. Also, a new line was added to include polarizability for the newly added atom type "o-".
- tleap -s -f leap.in This requires MOL.frcmod, MOL.mol2, MOL.pdb.
- parmed -i addc4_PO4.in. This requires resultPO4-nocount5.prmtop, c4.txt, pol_addc4_phosphate.dat.
- Go to the end of "resultPO4-nocount5_addc4.prmtop", and change "4.83448753E+01" to "0.00100000E+00". This is to fulfill criteria #2 but not to alter the energy.
- Still, at the end of "resultPO4-nocount5_addc4.prmtop", change "-2.35783788E+02" to "-1.17891894E+02". This is to fulfill criteria #3.
pmemd.cuda -O -i md_min1.in -o md_min1.out -p resultPO4-nocount5_addc4.prmtop -c resultPO4-nocount5.inpcrd -r md_min1.rst -x md_min1.netcdf -inf md_min1.mdinfo -ref resultPO4-nocount5.inpcrd
- Check whether the C4 matrix looks like Figure 1.2. Also, the first step of minimization should give a VDW energy of about 5884 kcal/mol.
Automated by Python:
To facilitate user manipulation of the C4 matrix generated by ParmEd, we have prepared a Python script named c4_replacer.py. This script is suitable for use in more complex systems than this Ca2+ and phosphate example.
As previously mentioned, this script can help automatically divide the C4 term between the phosphate oxygen atom and the metal ion by two. Additionally, this script can also replace any zero values between (o-)<->(o-) and (Ca2+)<->(Ca2+) with small numerical values to fulfill criteria #2.
To add or modify the C4 terms to the topology file using ParmEd, follow these steps to run the c4_replacer.py script:
- Make sure that ParmEd is installed in AMBER, or by using either “pip install parmed” or “conda install -c conda-forge parmed.”
- Place the following files in the same directory: c4_replacer.py, pol_addc4_phosphate.dat, resultPO4-nocount5_addc4.prmtop, c4.txt, and addc4_PO4.in.
- Run the script as follows, which will prompt to manipulate C4 terms. When prompted, specify the atom type (e.g., "o-") for which the C4 term needs to be changed.
python c4_replacer.py -i pol_addc4_phosphate.dat -p resultPO4-nocount5_addc4.prmtop -c addc4_PO4.in
Note: The script will create backup files with an 'old_' prefix for tracking changes to each file.- Enter the new polarizability value. Since the default polarizability value for the "Ca2+" and "o-" atom types in the pol_addc4_phosphate.dat file is 3.9 and XXX respectively. By updating the polarizability values to 1.95 and XXX respectively, ParmEd will then update the C4 term between "Ca2+" and "o-" to this new value -117.8 (half of -235.7). Up to this point, criteria #2 is satisfied.
- The script will inquire whether the user wants to manipulate another C4 term. Input "Ca2+" to handle the C4 term between Ca2+ ions, and enter a small value such as 0.001. ParmEd will set this value as the c4 term between the Ca2+_Ca2+. 6. Next, you need to set a small value between o- and o-. However, since it shares the same atom type with Ca2+_o-, you should enter 'n' to skip this step and make the change in step 7.
- In this step, the script will ask if you want to replace another c4 term in the matrix. Here, you can input the c4 terms between o- and o- (2.41724377E+01). And replace it with a suitable value, such as 0.00100000E+00.
- Finally, the script will display the updated c4 matrix of the system, resembling the figure mentioned earlier. The topology file is now ready for simulations.
You can use this script for different or more complex systems, but ensure you thoroughly understand the explanations provided above. If you want to use your own input files with the script, make sure to update the input file names in the addc4_man.in file.
Below is a special section for Charmm-GUI users, as well as users who are familiar with default 12-6-4 but want to explore modified 12-6-4 in AMBER:
When using Charmm-GUI to construct a simulation system for metalloproteins and metal transporters with C4 terms, it places the default C4 terms between the metal ion and the ligand chelating atom (e.g., the nitrogen atom of a histidine residue). To improve the accuracy of these interactions, it is necessary to replace these default C4 terms with finely tuned parameters as outlined in previous studies. The initial step is to use parmed to incorporate the modified C4 terms between the ligand's chelating atom and the metal ion.
This procedure closely mirrors the one previously described, but with the utilization of specific files: cg-addc4.in, cg-lj.dat, and c4-metal-ion.txt. These files modify the C4 terms between the zinc ion and the NE2 atom of HID residues in the protein. You can adjust the parameters, atom types, and metal ions according to the specific requirements of your simulation systems(Please read the contents of the cg-addc4.in file for more details).
It is crucial to specify a non-zero value in the cg-lj.dat file for the metal ion you are using when employing parmed. Therefore, while following the aforementioned process to integrate the modified C4 terms into your system, ensure that a non-zero value is present for the metal ion in the cg-lj.dat file referred to in the addc4.in file. This action prompts parmed to establish a non-zero C4 term between metal ion-metal ion, thereby enabling AMBER to activate C4 terms between the ligand's chelating atom and the metal ion in your system. You can apply this procedure to systems created without Charmm-GUI, but first, make sure you add the C4 terms between metal-water as described on the ambermd.org website. This flexibility allows for consistent implementation of the enhanced C4 terms in various simulation systems. To verify the successful activation of C4 terms, it is advisable to monitor the VDW energy of the system, as previously described, to ensure their effective incorporation into your simulation.
Conclusion
Having completed the first 2 steps of Figure 1.1, you are ready to make topology (prmtop) and coordinate (inpcrd) files using LEaP. For more information on topology and coordinate files as well as the flow of information in Amber, please read through section 1.1 on page 13 of the Amber 2020 Manual.To do so, you will have to load a force field which describes the potential energy of the molecules in your system. Different force fields are used for different types of molecules (e.g., DNA, RNA, proteins, lipids, carbohydrates). To learn more about force fields, please refer to section 3 on pages 35 through 62 of the Amber 2020 Manual.
For more information about building systems with LEaP, go back to the Building Systems tutorial page and find information relevant for your system.
By Zhen Li and Majid Jafari