|
|
Building Protein Systems in Explicit Solvent
By Abigail Held1 and Maria Nagan2
1from Bill Miller III's lab, Truman State University, 2Stony Brook University
Table of Contents
I.Introduction
II. Use VMD Visualize Software to Examine a Protein Structure
III. Evaluate and Analyze the Protein Structure to be Imported into Amber
A) Non-Standard Residues
B) Metals
C) Experimental methods noted in the paper associated with the PDB
D) Solvent molecules or crystallization buffer
E) Missing electron density (amino acids)
F) Disulfide Bonds
G) Protonation States
IV. Use LEaP to Build a Protein System in Explicit Solvent
References
Learning Outcomes
Use VMD to examine a protein structure.
Evaluate and analyze the protein structure to be imported into Amber.
Use LEaP to build a protein system in explicit solvent.
I. Introduction
This tutorial will go over how to build a protein system using AmberTools.
It is targeted toward beginners; however it may also be useful to
more experienced users who are building a protein system for the first
time. This tutorial requires AmberTools and VMD be properly installed
on a working Linux/Unix computer.
Process
II. Use VMD Visualization Software to Examine the Protein Structure
AmberTools is used through Unix command line, which is accessed via a
terminal window. Open a terminal now on
your computer. For help learning the command line in Unix, see:
1. Make a directory for your tutorial.
[Username@computer ~]$ mkdir tutorial
[Username@computer ~]$ cd tutorial/
To see the path to your working directory, you can use
pwd (print working directory).
[Username@computer tutorial] pwd
~/Username/tutorial/
2. Download a pdb file from the PDB Databank.
The Protein Data Bank (PDB), found at https://rcsb.org, is a central repository of experimentally
determined structures. A PDB is a file format that contains 3D coordinates for a biomolecule that were determined using an experimental method such
as X-ray crystallography, cryo-EM, or NMR spectroscopy. Each PDB has a unique code, called a PDBID, that allows it to be found and referenced quickly.
When selecting a PDB for your protein of interest, do not select the first PDB that you see. Consider the resolution, the sequence, and the presence
(or absence) of inhibitors, cofactors, or substrates. Several studies solve mutated proteins, or solve it in complex with small molecules. Consider
what it is that you are trying to model and choose a PDB accordingly.
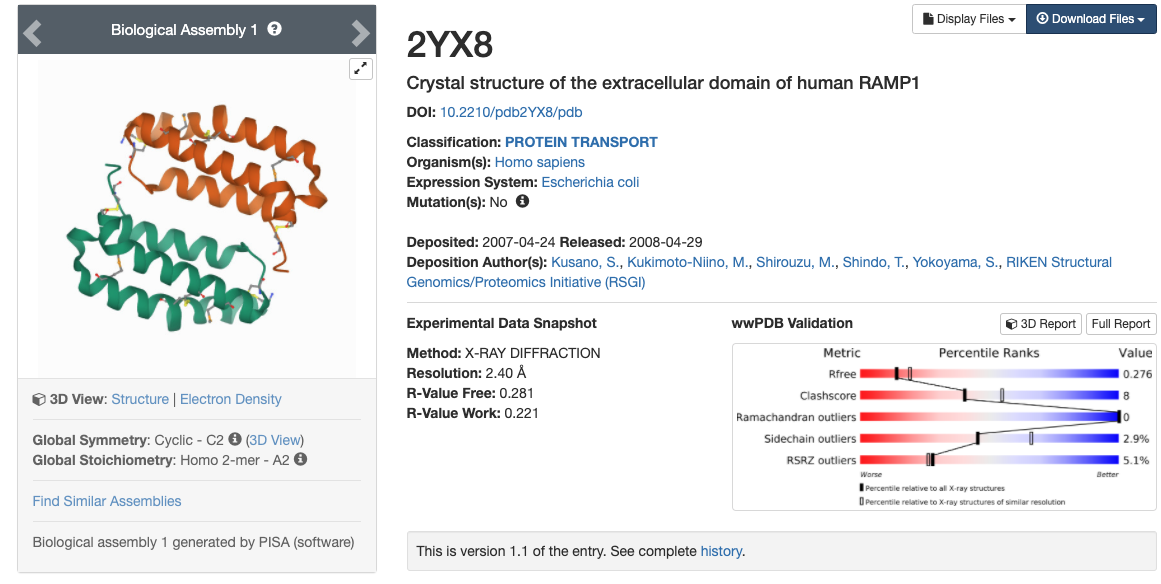
In this tutorial, we will be using the x-ray crystal structure of the human RAMP1 extracellular domain (PDBID: 2YX8).1 Go to the
protein data bank and download the PDB with the PDBID 2YX8.
- Enter 2YX8 into the search bar. You will be brought to a page with information such as the reference, a picture
of the structure, the sequence, and other information.
- You will be brought to a page with information such as the reference, a picture
of the structure, the sequence, and other information. Download the PDB file by clicking on the blue "Download Files" icon in the upper right hand corner and choosing "PDB Format".
This PDB file contains the coordinates for the extracellular domain of human RAMP1, and will be called 2yx8.pdb.
3. Move 2yx8.pdb into your tutorial directory.
Your web browser probably has a default location where it places downloaded files, such as
"Downloads".
[Username@computer]$ mv ~/username/Downloads/2yx8.pdb ~/username/tutorial/
4.Open VMD and load 2yx8.pdb into VMD.
The first thing you should always do when beginning to prepare a PDB is visualize it using a visualization software. Here, we use VMD, the
tutorials for which can be found under VMD Visualization Software if you are unfamiliar. You can
also use Chimera-X, the tutorials for which can be found under Chimera-X.
- Open VMD.
- Choose Select File, New Molecule, choose your 2yx8.pdb file, and select the file type: PDB.
- You can also open your PDB directly on Windows and Linux by typing:
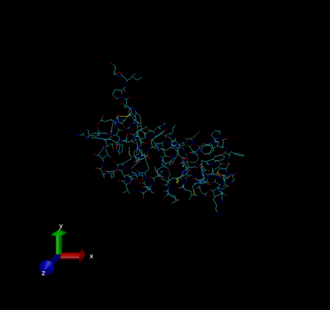
There will be three windows that open. The first is VMD main, which is basically your control panel. The second is the VMD terminal. The third is the
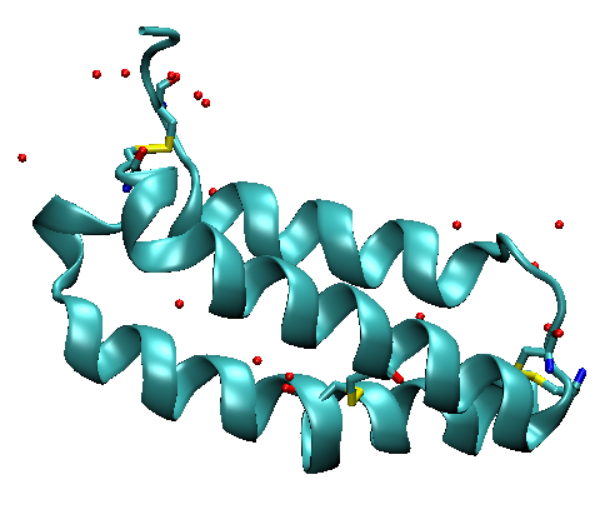
VMD display, which presently should look like this.
This display uses the default settings, which make the protein difficult to see. You can change aspects of this display to make it more useful. See the
VMD with AMBER tutorial for more information.
A few VMD functions
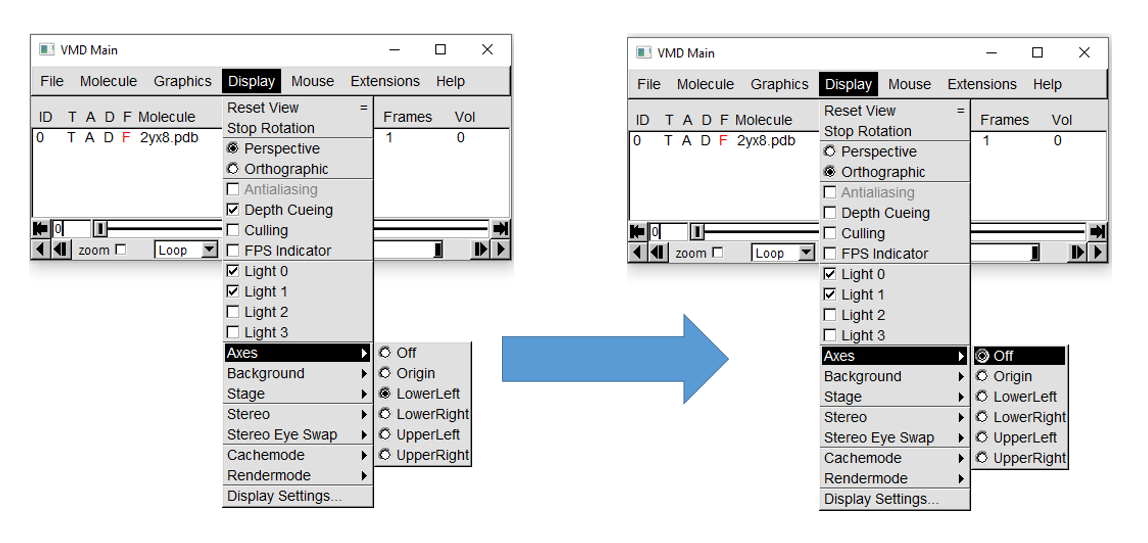
First, in VMD Main, hover the cursor over the "Display" tab. This will open a menu where you can alter these display settings.
For this tutorial, set the projection to orthographic, turn off depth cueing, and turn off axes.
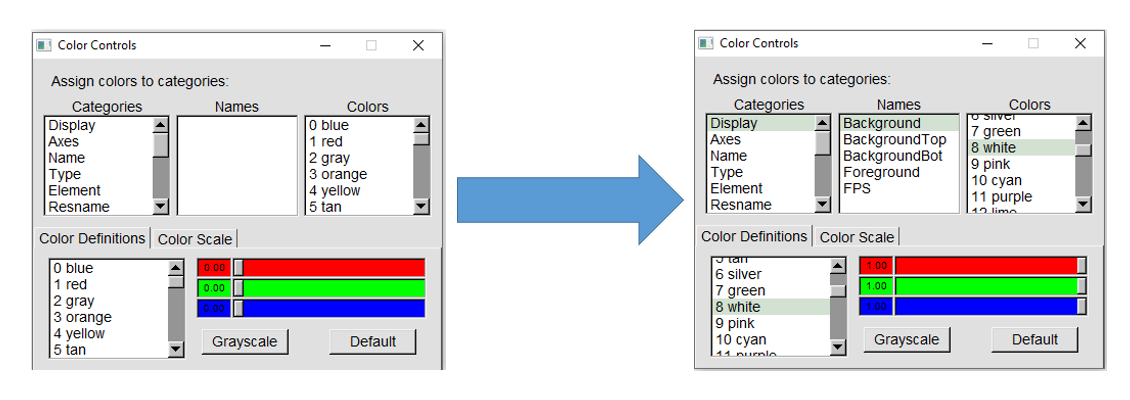
You can change the background color to white by hovering over the "Graphics" tab and clicking on "Colors...". This will open the Color Controls
menu. Choose "Display" in the "Categories" menu, "Background" under "Names", and "8 White" under "Colors," as shown below.
This will automatically change the background color to white in the Display window.
To more clearly see the protein itself, you can alter aspects about the protein display through the "Graphical Representations" menu under the
"Graphics" tab on the VMD Main window. This menu allows you to control aspects of the PDB display by creating new representations ("Create Rep")
and altering the drawing method, coloring method, and material. Here, we should make the protein visible as a backbone and the water
molecules more prominent. This protein also contains disulfide bonds, so we should make all cysteine residues visible. Make the following
representations in the "Graphical Representations" menu.
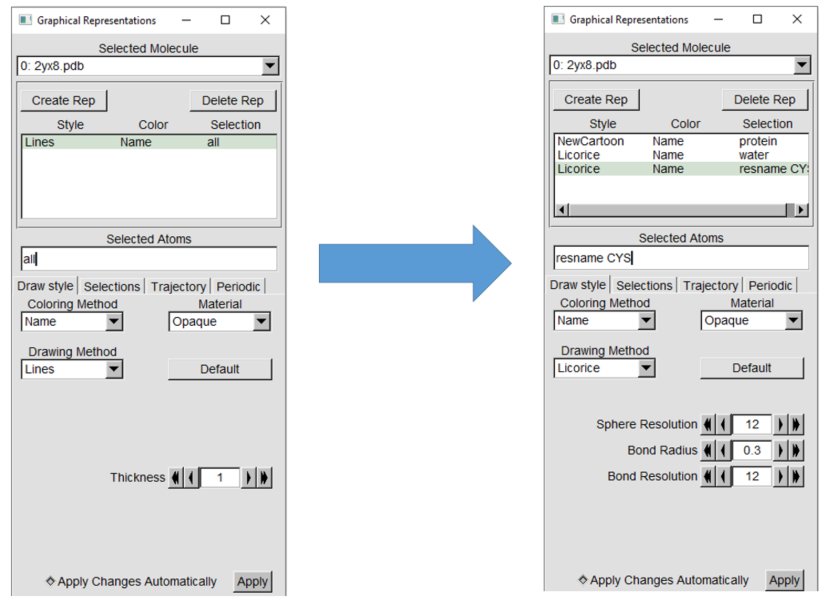
Use the new cartoon drawing
method for the protein and the licorice drawing method for water and cysteine. The display should have the changes applied automatically, but if not
click "Apply" at the bottom of the "Graphical Representations" window. The PDB that we are using for this tutorial does not contain any
non-standard residues, but if you are following this tutorial with a different PDB that does contain non-standard residues, you should show them.
Your display should now look something like this.
III. Evaluate and Analyze the Protein Structure to be Imported into Amber
You may have noticed that the PDB does not contain any hydrogens. This is because X-ray methods cannot resolve hydrogens, so they are not included
in X-ray structures.
2 Do not worry about this, as hydrogens will be
added later.
PDBs are never ready for use in MD simulations as they come! They require several modifications before they can be
used. Below is a checklist of what to look for in your protein PDB before using it for MD simulation. The procedure for dealing with items marked
with a * will be covered in this tutorial. Anything not marked is beyond the scope of a beginning tutorial, though they should still be considered.
A) Non-Standard Residues
B) Metals
C) Experimental methods noted in the paper associated with the PDB
D) Solvent molecules or crystallization buffer
E) Missing electron density (amino acids)
F) Disulfide Bonds
G) Protonation States
A) Non-standard Residues
Non-standard residues refer to any residue in your PDB that is not a standard amino acid. These include cofactors
( NADH, heme, etc.), non-standard amino acids (hydroxyproline,etc.), and bound inhibitors or substrates. If you have them, you should consider how you
want to model these residues. In the case of small organic molecules that do not contain metals or metalloids, this could be as
simple as using antechamber (see Tutorial 2.1 Simulating a pharmaceutical compound using antechamber and the Generalized Amber Force Field).
In the case of more complicated non-standard residues, you may have to use more advanced methods.
B) Metals
Dealing with metals can be very complex, and you should carefully consider how to model them if you have one. There are parameters for monovalent
and divalent ions listed on the ions force fields page , in the Amber Manual in Section 3.6 as well as in $AMBERHOME/dat/leap/lib and $AMBERHOME/dat/leap/parm.
C) Experimental methods noted in the paper associated with the PDB*
You should always read the paper associated with your PDB. Read the 2YX8 paper by Yokoyama and coworkers and note any important structural features (i.e. disulfide bonds).
Also note the experimental procedure(s) used to solve the structure. Here, multi-wavelength anomalous dispersion (MAD) was used, which involves
replacing methionine residues with selenomethionine. We will need to edit the PDB file to fix this.
Remember that you are trying to simulate reality, which is not necessarily emulated in the PDB!
a. Make a copy of 2YX8 to perform the changes on with cp:
cp 2yx8.pdb 2yx8_fxMET.pdb
Each amino acid residue in the PDB file has a three letter residue name (resname) which corresponds to the three-letter code. Residues like
selenomethionine will have different residue names.
b. Open your
PDB file using vi or another text editor.
If you are unfamiliar with the use of
vi, see
this tutorial.
c. Look for the MODRES
In the PDB heater information right under the sequencing
data, 2yx8.pdb contains MODRES records that list selenomethionine (MSE) residues that need to be modified to methionine (Residue 48 and 76).
Look through the PDB for the first residue with the MSE resname listed in the MODRES records. It should look like this.
d. Clean up the file and replace MSE with MET
- The HETATM tag in the first column means that these residues will not be connected to the surrounding amino acids
via a bond by default.
Find/replace HETATM to "ATOM" so that the sequence will be continuous. Be sure to keep the two spaces after the ATOM so the rest of the columns will
be lined up.
- The second column is the atom number. Do not edit this!
- The third column is the name of the atom. For example, CA is the alpha carbon. Because methionine does not contain
selenium, we need to change
this atom to sulfur. Edit the atom designation from SE to SD (the atom name of the sulfur atom in methionine). Change the SE in the last column to
an S as well.
- The fourth column is the resname that was mentioned above. Change all of the MSE entries to MET.
After making these changes, you should have this.
- Make the same changes to the other Met residue listed in the MODRES records and save your changes.
D) Solvent molecules or crystallization buffer*
On several occasions, solvent or crystallization buffer may have been crystallized along with the protein and are included in the PDB. In 2yx8, for
example there are several crystallographic waters. Because we are preparing an explicit solvent system, we can keep these. Sometimes, other solvents or
phosphates will be present. These do not affect the function of the protein, so they can be removed.
E) Missing Electron Density (amino acids)
Sometimes, the researchers who solved a PDB could not get a clear enough picture of certain amino acids. This means that the PDB is missing electron
density, and you have to model in the missing residues. To check if you have this problem:
- You can use VMD to show the protein backbone (using new cartoon; see Learning Outcome 1) and look for any gaps.
- Alternatively, you can look in the PDB file itself for missing residues. If you are missing residues 15-18, for example, you will see information
on resid 14 and then resid 19.
If you find any gaps, you will need to seek out methods for modelling in your missing amino acids.
Check 2yx8_fxMET.pdb for missing electron density
using either (or both) of these methods.
F) Check for disulfide bonds*
We read the paper and saw the protein in VMD (Learning Outcome 1), so we
know that this protein contains disulfide bonds. Cysteines involved in disulfide bridges have a special Amber resname (CYX) however these will not be
in the PDB you got from the Protein Data Bank.
a. Find the cysteines in the disulfide bond.
You can find the cysteines involved in disulfide bridges listed in the SSBOND records near the top of
the PDB.
Each of these cysteine residues must have their CYS resnames changed to CYX in your PDB. This is just like how we edited MSE to MET above, except it's
less complicated because you only need to change the resname.
b. Make a new copy of your PDB with a _fxCYS tag.
cp 2yx8_fxMET.pdb 2yx8_fxMET_fxCYS.pdb
c. Use a text editor (like vi) to change residues named "CYS" involved in a disulfide bond to "CYX".
We are not entirely done dealing with disulfide bonds yet, however this is all we can do until we use LEaP to build the system ( Learning Outcome 3).
#7) Check protonation states*
Remember that our end goal is to simulate reality, and that may not necessarily be reflected in the PDB. One of the big examples of this is protonation
states. Several proteins contain amino acids with non-standard protonation states. For example, aspartate proteases have a protonated Asp residue in
their active site. You should know your protein and how it functions before you try to simulate it, and you should know if it requires any non-standard
protonation states. If so, you will have to edit your PDB.
Protein PDBs that were solved with an X-ray method do not contain hydrogens because the method is not capable of resolving them. LEaP automatically
adds hydrogens to these PDBs based off of optimal hydrogen bonding while following standard protonation states. Therefore, the amino acid with a
non-standard protonation state will get protonated incorrectly if you do not make the necessary resname change(s). For example, in a PDB for an Asp
protease, the protonated Asp will come with the resname ASP, just like all the other aspartates that are carboxylates. This will cause LEaP to protonate
it as if it were a regular aspartate. To prevent this, you would have to change your non-standard Asp's resname to ASH (the resname for protonated
Asp). With the correct resname, LEaP will correctly protonate your amino acids. Table 1 shows the resnames for some common protonation states.
Table 1. AMBER Resnames of Common Non-standard Protonation States
Non-standard Protonation Form
|
AMBER Resname
|
Protonated/uncharged Asp
|
ASH
|
Protonated/uncharged Glu
|
GLH
|
Deprotonated/uncharged Lys
|
LYN
|
His protonated at epsilon position
|
HIE
|
His protonated at delta position
|
HID
|
Charged His (protonated at both positions)
|
HIP
|
Deprotonated Cys or Cys bound to a metal
|
CYM
|
Cys involved in disulfide bridge
|
CYX
|
You may have noticed that the three resnames for histidine given in Table 1 are the only possible protonation states for histidine. LEaP will convert
all residues named HIS in the PDB to HIE (epsilon protonated) if it is not specified according to the His resnames in Table 1. The problem with this is
that the epsilon position may not always be the optimal position for hydrogen bonding.
The program H++3 can predict the protonation states of histidine if given a PDB.
a. Go to the H++ website (above)
and click on "process a structure".
You can search for your protein PDB directly from the database, but this will result in a failed calculation
because of the HETATM designation on the selenomethionine residues.
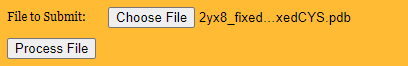
b. Upload your PDB with the _fxMET_fxCYS tag
See below, and click
"process file."
c. Choose the correct pH.
You will see various parameters for running your calculation. The experiment to solve the structure of 2YX8 was conducted at pH 5.4. We want to run our
simulation at biological pH, so change the pH from 5.4 to 7.0, as shown below.
d. Run the H++ calculation
- Click the "process" arrow on the bottom right and wait for the calculation to complete.
- Click "view results".
- Note the charge of your protein because we will need to know it later when we neutralize the charge of the system. The charge of
2YX8 is -2.
e. Download the new pdb file.
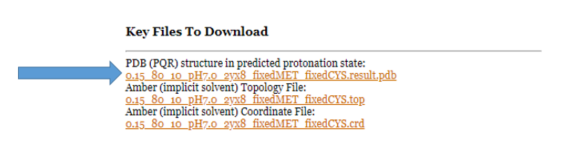
You will be able to download the PDB structure with the predicted protonation states. Do this now by clicking on
"0.15_80_10_pH7.0_2yx8_fxMET_fxCYS.result.pdb" under "Key Files to Download."
e. Look at the new pdb file from H++ with VMD.
- Move the file to your tutorial directory, load it into VMD, and show each histidine residue (resname HIS).
- Click on the VMD display and press 1. Your cursor will turn into crosshairs, and you can click on a residue to show the
resname, resid, and atom name. It should look similar to this.
You can see that His75 is protonated at both positions (HIP), and His97 is protonated at the delta position (HID). If you are unfamiliar with the
histidine protonation states, take some time to Google it.
f. Make a copy of your 2yx8_fxMET_fxCYS.pdb with an added _fxHIS tag.
cp 2yx8_fxMET_fxCYS.pdb 2yx8_fxMET_fxCYS_fxHIS.pdb
g. Edit the pdb file to contain the correct residue names for HIS.
- Open your _fxHIS file and edit the resname of His75 to HIP and His97 to HID, in accordance with the resnames given in Table 1.
- If you are following this tutorial with a different protein and one of your histidines is directly involved in the protein's function
(such as it is an active site residue), then you should see if you can find the protonation state in the literature. If not, then go with H++'s
prediction.
h. Remove CONECT from the pdb file.
The last thing you should do is remove the CONECT records at the end of the PDB. These describe the bonds that certain atoms have to each other, and
they often confuse LEaP (Learning Outcome 3). Fortunately, the CONECT records are not necessary for proper MD simulation, so they can be deleted out
of your PDB.
IV. Use LEaP to Build a Protein System in Explicit Solvent
A functioning protein system is described in two files: the topology (.prmtop) and initial coordinates (.inpcrd). See the
Fundamentals of LEaP tutorial for more information on specific file types. LEaP
is a tool that we use to turn our PDB into a prmtop and inpcrd. LEaP is also what we use to explicitly solvate the protein, neutralize the charge, and
add an appropriate salt buffer.
a. Set AMBERHOME
In order to run an executable in Amber, you will need to first define the
AMBERHOME environment variable.
The installation step will create a resource file, e.g., amber.sh ,
in your installation directory. This script will set up your shell
environment correctly for Amber, e.g.: <\p>
source /home/xxxx/amber20/amber.sh # for bash, zsh, ksh, sh, etc.
Adding these commands to your login resource file (e.g., ~/.bashrc, ~/.zshrc,
etc.) will set up your environment every time you start a new shell.
[There is a similar script, amber.csh , for those who use a C type
shell interactively.]
It is also possible to define AMBERHOME manually, but this is not recommended
because some executables require other environment variables.
b. Start tleap
LEaP functions through an interface called
tleap
or
c. Load force fields
- The choice of force field is very important. If you are simulating proteins, for example, you do not want to use force fields for RNA.
Furthermore, there are several water models to choose from (OPC, TIP3P, and TIP4P are some common examples), and they each have a set of parameters
for ions that are meant to work with that water model. So, you can't use the OPC water model and the TIP3P ions force field. In short, the force
fields that you choose have to match what you are simulating and work with each other.
- If your system contains non-standard residues, you will have to add in additional parameters that describe those. Here, we will only have
protein, water, and ions to worry about, so this will not be covered.
See the AMBER Manual for more information on force fields.
- In this tutorial, we will be using the FF19SB force field for proteins,4
the OPC water model,5 and the matching
Li-Merz 12-6 ions model6.
- The first line loads in our protein force field using the ff19SB force field. This force field is the most recently updated AMBER force field
for proteins, but if you do not have a newer version of Amber you may use ff14SB. The third line loads in the OPC water model and the 12-6 ions for OPC.
source leaprc.protein.ff19SB
source leaprc.water.opc
d. Load the pdb file into a UNIT called "ramp"
Here, we define a variable for our PDB. What this means is that any time we say "ramp" in a tleap script or in this interactive session of tleap, the program will know that we are
referring to RAMP1.pdb, and will impose any actions we perform onto RAMP1.pdb. The variable can be anything you want as long as you are consistent.
e. Form disulfide bonds between CYX residues
The CYX resname for disulfides only tells LEaP that a Cys is involved in a disulfide bridge, not which cysteines are bound to which. That is what we
define next. The bond command tells LEaP that we are forming a bond between these two specific atoms. Make sure that you are bonding the correct atoms
to each other! Use the SSBOND records to keep track of which cysteines are bound to which.
In interactive mode, type:
bond ramp.27.SG ramp.82.SG
bond ramp.40.SG ramp.72.SG
bond ramp.57.SG ramp.104.SG
f. Save a prmtop and inpcrd files of just the protein for analysis later.
saveAmberParm ramp RAMP1_gas.prmtop RAMP1_gas.inpcrd
g. Add neutralizing charges
Next, we neutralize the charge of our system by adding counter ions. We will be using NaCl as our salt buffer, so we can use Na+ and Cl- as our counter
ions. When you ran H++ in Learning Outcome 2, you took note of the charge of the protein. For 2YX8, it was -2, so we will be using 2 Na+ atoms to
balance the charge.
In interactive mode, type:
This will add 2 Na+ ions to the system. If, for whatever reason, you did not run H++ on your protein and you do not know the charge, you can
replace the 2 with a 0 to tell LEaP to neutralize
the system with your selected ion. The problem with this is that if your charge is -2.99999, LEaP will only add 2 counter ions even though there should
be 3. If you know the charge of your protein, it's best to add in a specified amount.
h. Add a truncated octahedron box of OPC water
Next, we solvate our system in explicit solvent.
SolvateOct tells LEaP to solvate RAMP1.pdb (with all the cysteine bond modifications and added counter ions) in a box shaped like a truncated octahedron.
This shape mimics a sphere, and is a very common box shape to use. The number tells LEaP how big to make the box. Here, every part of the protein will be
at least 10.0 Å away from the edge of our water filled box.
Generally, you want at least three layer of solvation7 on all sides of the protein surface for an MD simulation, and 10.0 Å will get you that.
In interactive mode, type:
solvateOct ramp OPCBOX 10.0
i. Add a buffer ions with random positions.
Next, we need to add a 150 mM NaCl salt buffer that is present in cells, which will require adding Na+ and Cl- ions, based on
the volume of the box. Your tleap window should have the following information. The same information can also be found in the leap.log file. The volume is hightlighted in blue below.
Scaling up box by a factor of 1.413369 to meet diagonal cut criterion
Solute vdw bounding box: 45.400 31.108 30.346
Total bounding box for atom centers: 73.667 73.667 73.667
(box expansion for 'iso' is 55.9%)
Solvent unit box: 18.865 18.478 19.006
The number of boxes: x= 4 y= 4 z= 4
Volume: 208141.839 A^3 (oct)
Total mass 110188.212 amu, Density 0.879 g/cc
Added 5569 residues.
Calculate how many Na+ and
Cl- ions are required to have this concentration with this box volume.
See the 1.5 Calculating Salt Molarity Tutorial if you need help.
You should have gotten that 19 Cl- ions and 19 Na+ ions are required for an initial 150 mM NaCl buffer concentration in this box.
Add these ions in randomized positions.
addIonsRand ramp Na+ 19 Cl- 19
j. Save the prmtop and inpcrd files
saveamberparm ramp RAMP1_ion.prmtop RAMP1_ion.inpcrd
k. Check for tleap errors.
NOTE: As you execute the commands in tleap, you should get a message that describes both what was done
to the system (such as where ions were placed, the volume of the box, etc.) and how many
errors and warnings were output by LEaP, which should be none. Any Errors or Warnings should be examined to determine if they are consequential.
In particular, versions released after Amber 20 may emit warnings like this:
** Warning: No sp2 improper torsion term for C*-CN-CB-CA atoms are: CG CE2 CD2 CE3
These can be safely ignored.
l. To exit LEaP, type:
Next Steps
Now you should have a prmtop and inpcrd that together contain parameters and coordinates for our modified 2YX8 PDB, the OPC water model with a
sufficient box size, ions to neutralize the charge, and the salt buffer found in cells. Next
we should visualize it with VMD to make sure that everything that we added is actually there. This means that we need to check
that:
1. The protein backbone looks like a protein and doesn't have any missing sections
2. The correct disulfide bonds were added
3. The correct histidine protonation states were added
4. The water was added
5. The counter ions were added
6. The buffer looks reasonable
You can download the files from this tutorial:
Note that the position of the ions are random, so yours will
look different from these. If you are struggling, compare what you have to these files.
The next steps for our protein system would be relaxing the solvent, heating, and equilibration to further prepare the system before a production run.
Leap Scripts
tleap will also allow the user to read in a script containing a set of directions. The first thing to do is make a file for
our tleap script:
touch tleap.in
You can also use a text editor like vi to create a file called "tleap.in". In your tleap.in file, input the
following contents.
File: tleap.in
#tleap.in
source leaprc.protein.ff19SB
source leaprc.water.opc
loadamberparams frcmod.ions1lm_126_hfe_opc
ramp=loadpdb RAMP1.pdb
bond ramp.27.SG ramp.82.SG
bond ramp.40.SG ramp.72.SG
bond ramp.57.SG ramp.104.SG
SaveAmberParm ramp RAMP1_gas.prmtop RAMP1_gas.inpcrd
addIons ramp Na+ 2
solvateOct ramp OPCBOX 10.0
addIonsRand ramp Na+ 19 Cl- 19
SaveAmberParm ramp RAMP1_ion.prmtop RAMP1_ion.inpcrd
The # at the beginning indicates that it is a comment by the writer of the script, and the program does not actually read it. In general, putting
a descriptor at the top of tleap scripts is a good idea. The rest are the commands in the gray boxes above, are all the interactive commands from above compiled into one file.
To start tleap, read in the tleap.in file, and save a leap.log file (-f), type:
$AMBERHOME/bin/tleap -f tleap.in
Alternatively, you can also start tleap and source the tleap.in file:
$AMBERHOME/bin/tleap
source tleap.in
This will start tleap, execute your tleap.in script, and generate a leap.log file.
Use Cpptraj to make a PDB File
Make a pdb.in file that reads in your inpcrd file and makes a new pdb file.
trajin RAMP1_ion.inpcrd
trajout RAMP1_wions_water.pdb PDB
run
To execute:
$AMBERHOME/bin/cpptraj -p RAMP1_ion.prmtop -i pdb.in>pdb.out
Here is the pdb file made RAMP1_wions_water.pdb that you can load into VMD or another visualization program.
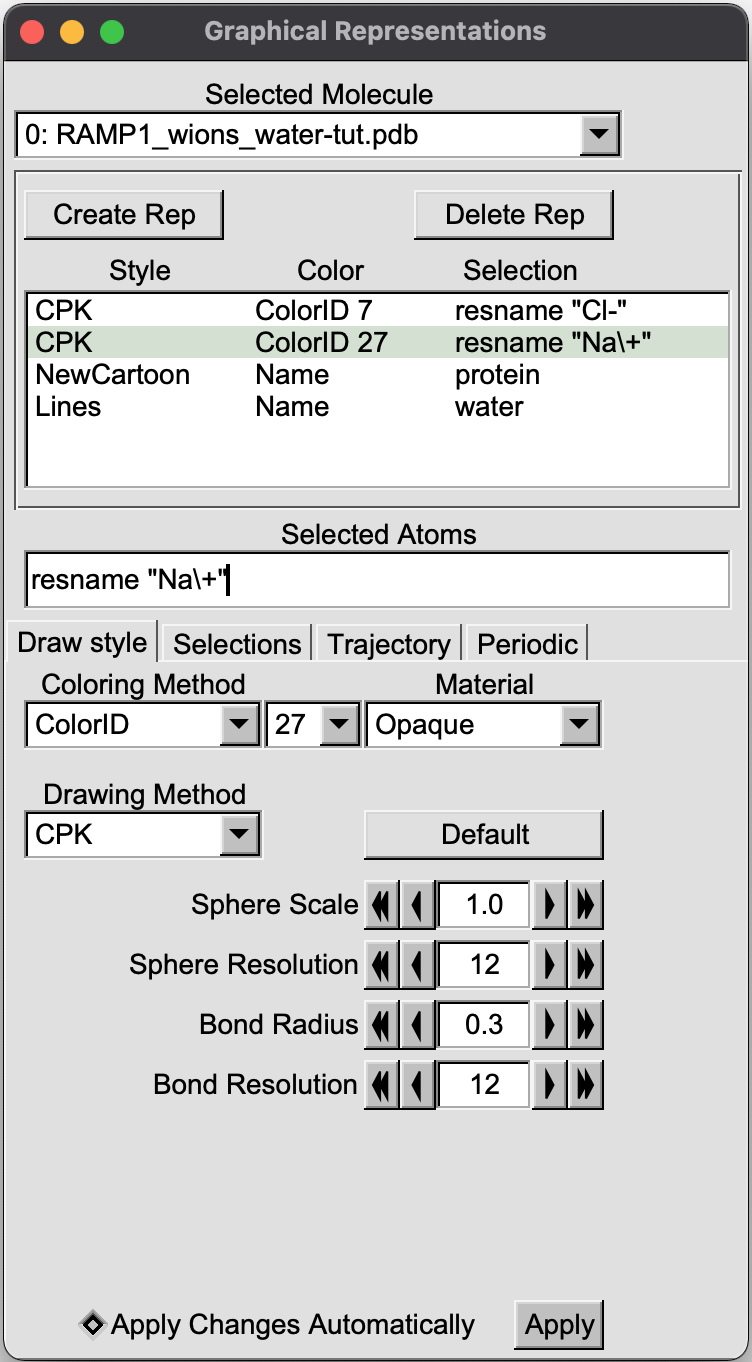
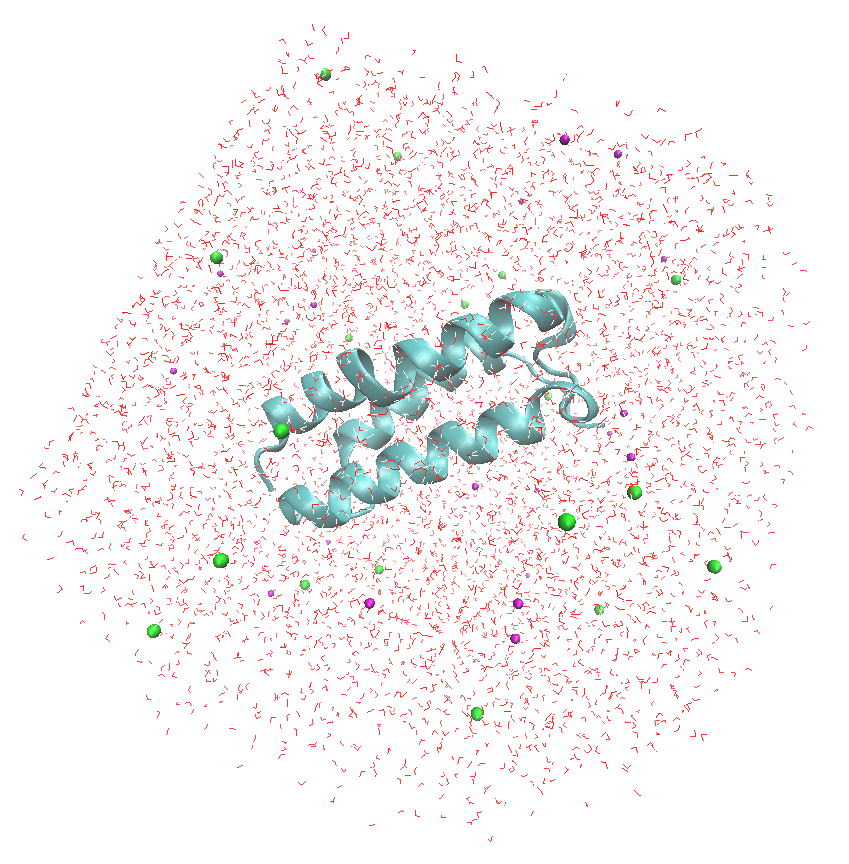
To better visualize the ions and water molecules around the protein, set the "Graphical Representations" as follows
The output in VMD will be displeyd as bellow:
References
1. Kusano,S.; Kukimoto-Niino,M.; Akasaka,R.; Toyama,M.; Terada,T.; Shirouzu,M.; Shindo,T. and S. Yokoyama "Crystal structure of the human receptor activity-modifying protein 1 extracellular domain."Protein Science, 2008, 17, 1907-1914. Comment: 2YX8.pdb
2. Elias,M.; Liebschner,D.; Koepke,J.; Lecomte,C.; Guillot,B.; Jelsch,C. and E. Chabriere "Hydrogen atoms in protein structures: high-resolution X-ray diffraction structure of the DFPase."BMC Research Notes, 2013, 6, 308. Comment: Hydrogens not in x-ray structures
3. Anadakrishnan,R.; Aguilar,B. and A.V. Onufriev "H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations."Nucleic Acids Research, 2012, 40(W1), W537-W541. Comment: H++ Program
4. Tian,C.; Kassavajhala,K.; Belfon,K.A.A.; Raguette,L.; Huang,H.; Migues,A.N.; Bickel,J.; Wang,Y.; Pincay,J.; Wu,Q. and C.Simmerling "ff19SB: amino-acid specific protein backbbone parameters trained against quantum mechanics energy surfaces in solution" J. Chem. Theory Comput., 2020, 16(1), 528-552. Comment: FF19SB Force Field
5. Izadi, S.; Anandakrishnan, R. and A.V. Onufriev "Building water models" J. Phys. Chem. Lett., 2014, 5(21), 3863-3871. Comment: OPC water
6. Li, P.; Song, L.K. and K. M. Merz, Jr. "Systematic parameterization of monovalent ions employing the nonbonded model"J. Chem. Theory Comput., 2015, 11(4), 1645-1657. Comment: HFE Ions for OPC water
7. Ebbinghaus,S.; Kim,S.J.; Heyden,M.; Yu,X.; Heugen,U.; Gruebele,M; Leitner,D.M. and M. Havenith "An extended dynamical hydration shell around proteins." PNAS,2007,104(52),20749-20752. Comment: layers of hydration
|
|
"How's that for maxed out?"
|
Last modified:
|
|
|
|