


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2013
Using Accelerated Molecular Dynamics (aMD) to enhance sampling
Formerly known as AMBER Advanced Tutorial 22
By Romelia Salomon, Levi Pierce & Ross Walker
In this tutorial we will learn how to harness the computational power of GPUs and the sampling power of accelerated Molecular Dynamics to efficiently discover long lived conformational transformations in the Bovine Pancreatic Trypsin Inhibitor (BPTI) protein. This tutorial closely follows the methods used in our paper Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira, J. Andrew McCammon, and Ross C. Walker. "Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics". J Chem Theory Comput. 2012 September 11; 8(9): 2997-3002. Conventional molecular dynamics allows one to access time scales on the order of tens to hundreds of nanoseconds; however, many biological processes of interest occur on longer time scales of up to milliseconds or more.
Accelerated Molecular Dynamics (aMD) is a bias potential introduced by the McCammon group at UCSD Hamelberg, Donald; Mongan, John; McCammon, J. Andrew. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys., 2004, 120(24), 11919-11929. It is a modification to the potential that in practice reduces the height of local barriers, allowing the calculation to evolve much faster. aMD represents an interesting option as it only requires the evolution of a single copy of the system, plus it doesn't require any previous knowledge of the shape of the potential.
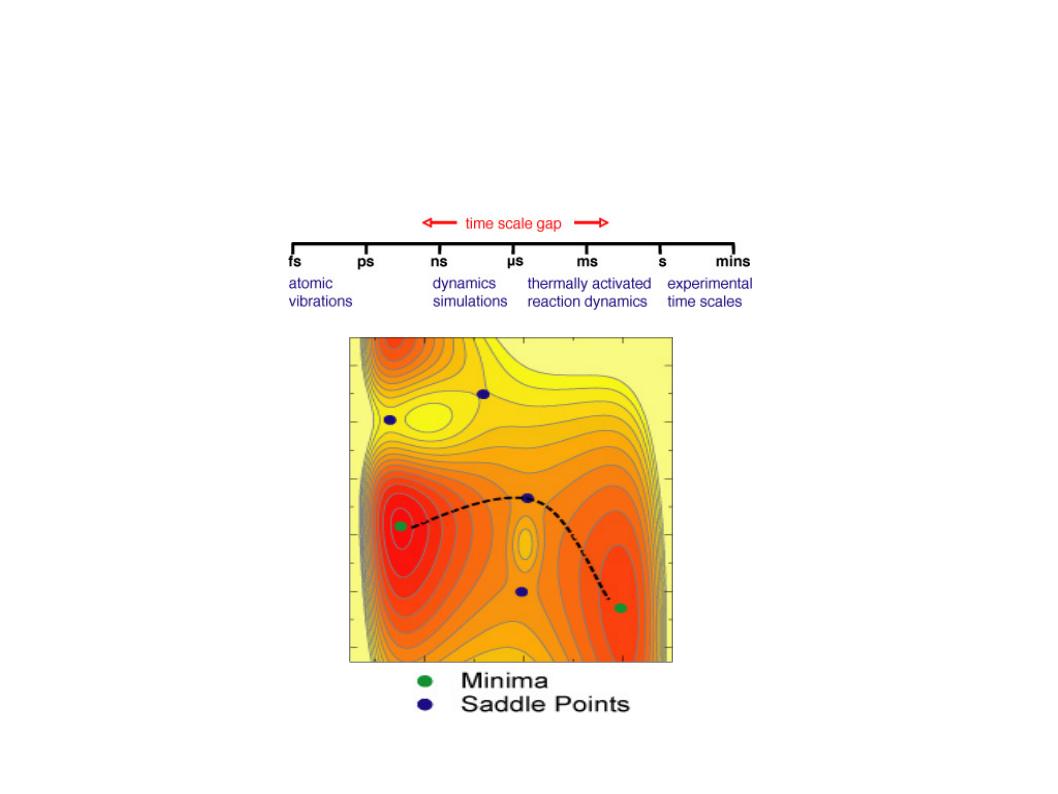
There are two main versions of aMD, one that raises the valleys and one that lowers the barriers. The later has only been introduced recently and its usability and benefits have not been fully studied. This method is commonly referred to as windowed aMD (w-aMD). In this tutorial we will use the original aMD formulation, however both versions are available now on AMBER. The modification of the potential required by the basic aMD boost is defined by the following equations:
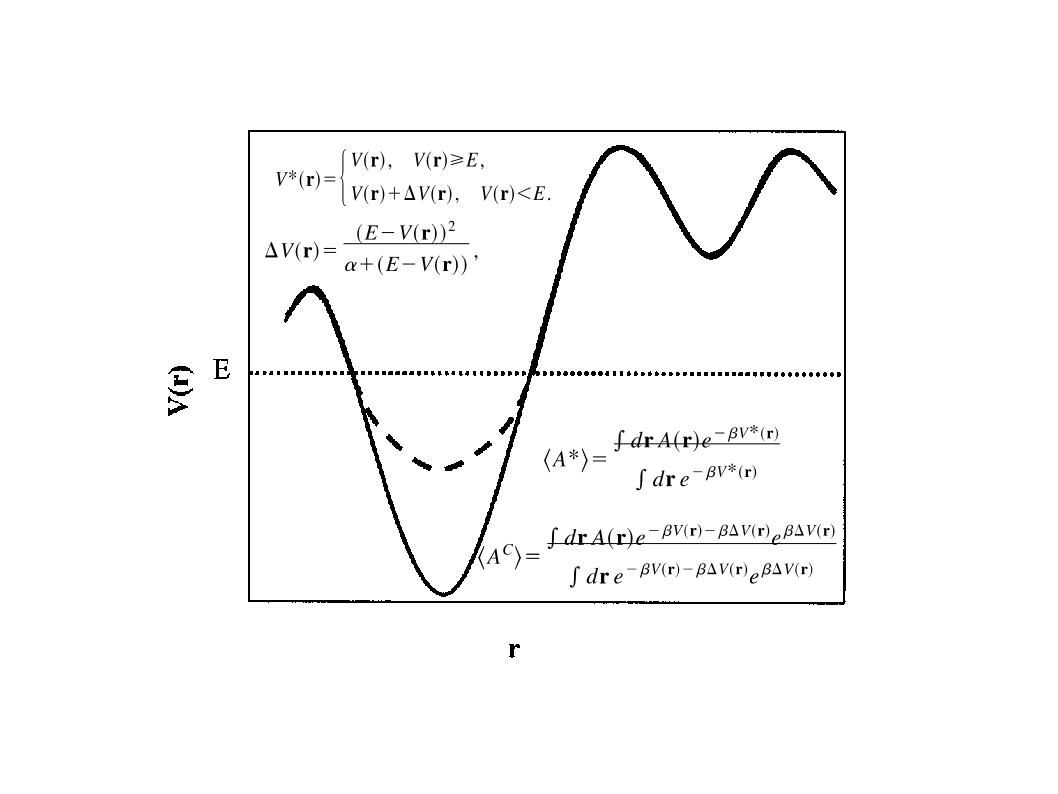
The present implementation of aMD in AMBER can work both lowering barriers and raising minima. AMD has been implemented in all AMBER MD engines (sander, pmemd and pmemd.cuda) by Romelia Salomon-Ferrer. The implementation includes the possibility of boosting independently only the torsional terms of the potential (iamd=2) or the whole potential at once (iamd=1). It also allows the possibility to boost the whole potential with an extra boost to the torsions(iamd=3). The only extra parameters needed to be specified in an aMD simulation are:
a) EthreshP. Average total potential energy threshold.
b) alphaP. Inverse strength boost factor for the total potential energy.
c) EthreshD. Average dihedral energy threshold.
d) alphaD. Inverse strength boost factor for the dihedral energy.
In this tutorial we will use the combined power of GPU acceleration, plus the sampling power of aMD raising minima to study conformational transitions that occur in BPTI on the microsecond time scale, comparing to a millisecond long conventional Molecular Dynamics simulation Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; Wriggers W. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341-346.
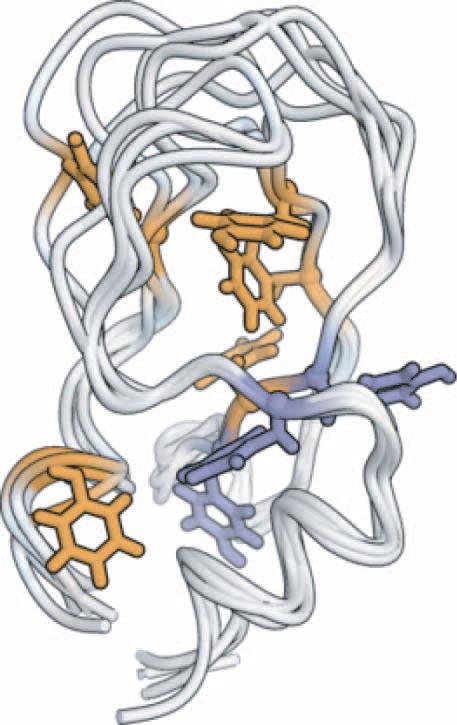
This tutorial consists of three sections:
1) section1 : Creating the initial structure and relaxing it.
2) section2 : Running the aMD calculations in Amber.
3) section3 : Data Analysis.
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2013