


Copyright (C) Pengfei Li 2016
Material systems modeling tutorial
By Pengfei Li
Formerly known as "AMBER Advanced Tutorial 27"
In this tutorial we will explore possibilities of employing the functionalities in the AMBER software package to model material related systems.
The AMBER software package has the state-of-the-art support for the molecular dynamics (MD) simulations, for example, the reliable and super fast MD engine - pmemd.cuda, very user-friendly parameterization program - antechamber, etc. However, its associated force fields are usually biologically related. Material systems are usually more diverse, and some force fields have been developed for them. Generally, there are inorganic, organic, and inorganic-organic mixed material systems. Some examples will be showed in this tutorial of modeling material related systems by using the AMBER software package together with the INTERFACE force field.[1]
A Protein System at FCC Ag Metal Surface
In this tutorial, we will use the INTERFACE force field developed by Heinz et al. [1] to model the {100} surface of the FCC Ag metal, and AMBER force field to model the small protein system.
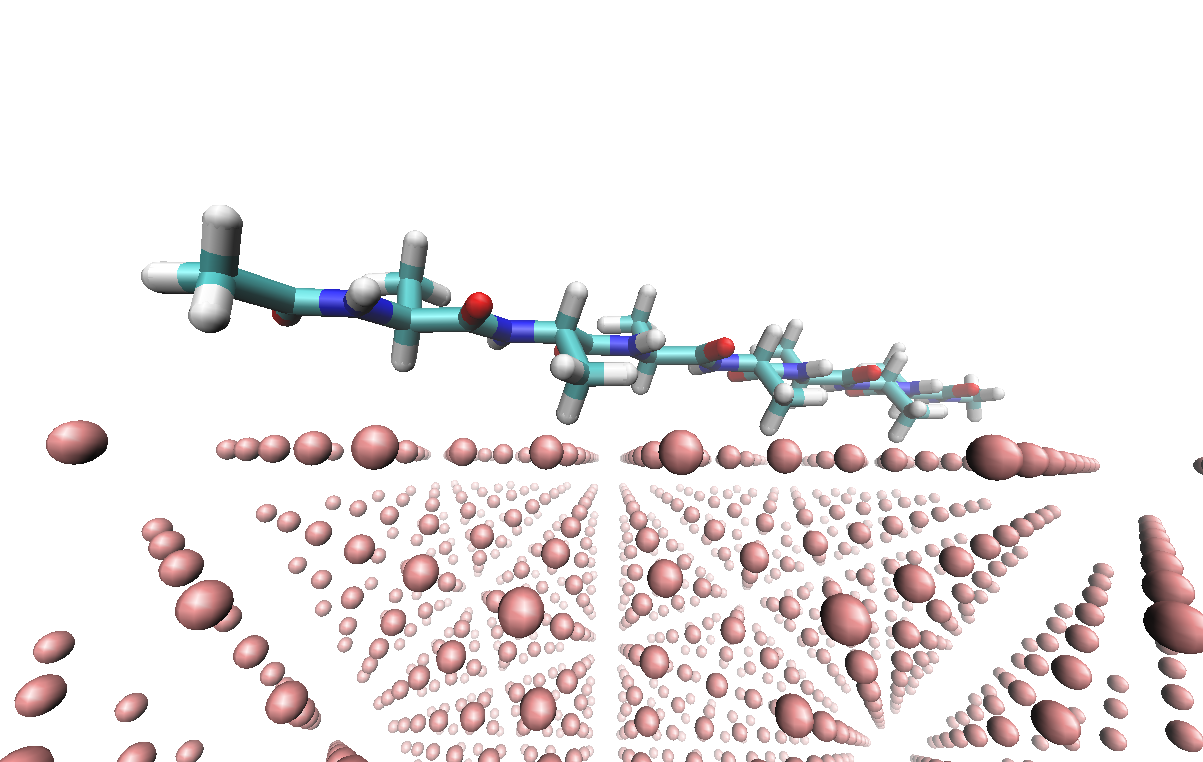
Click here to go to: Protein-Metal Surface Tutorial
The Polyethylene Terephthalate (PET) Polymer System
Organic polymer systems are very similar to the biopolymers have been parameterized in the AMBER force fields. Herein we will take advantage of the antechamber program and enjoy the convenience to model a PET polymer system.
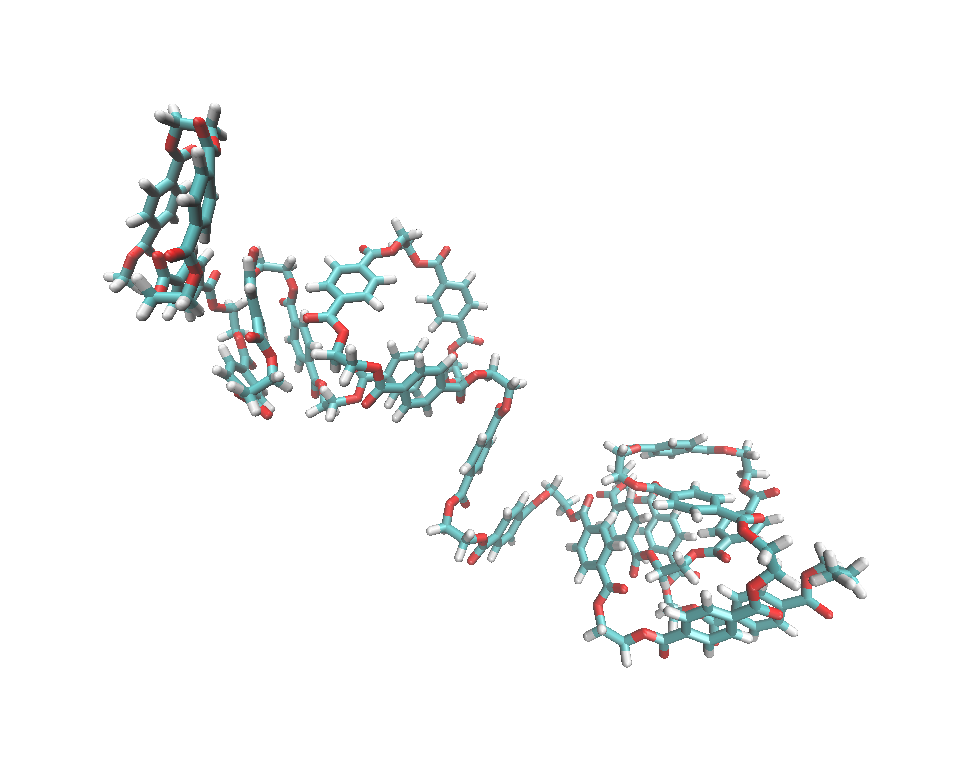
Click here to go to: PET Polymer Tutorial
A Hydroxyapatite-Water System
We will use the INTERFACE force field developed by Heinz et al. [1] for modeling the hydroxyapatite system, and SPC/E water model to model the liquid solvents.
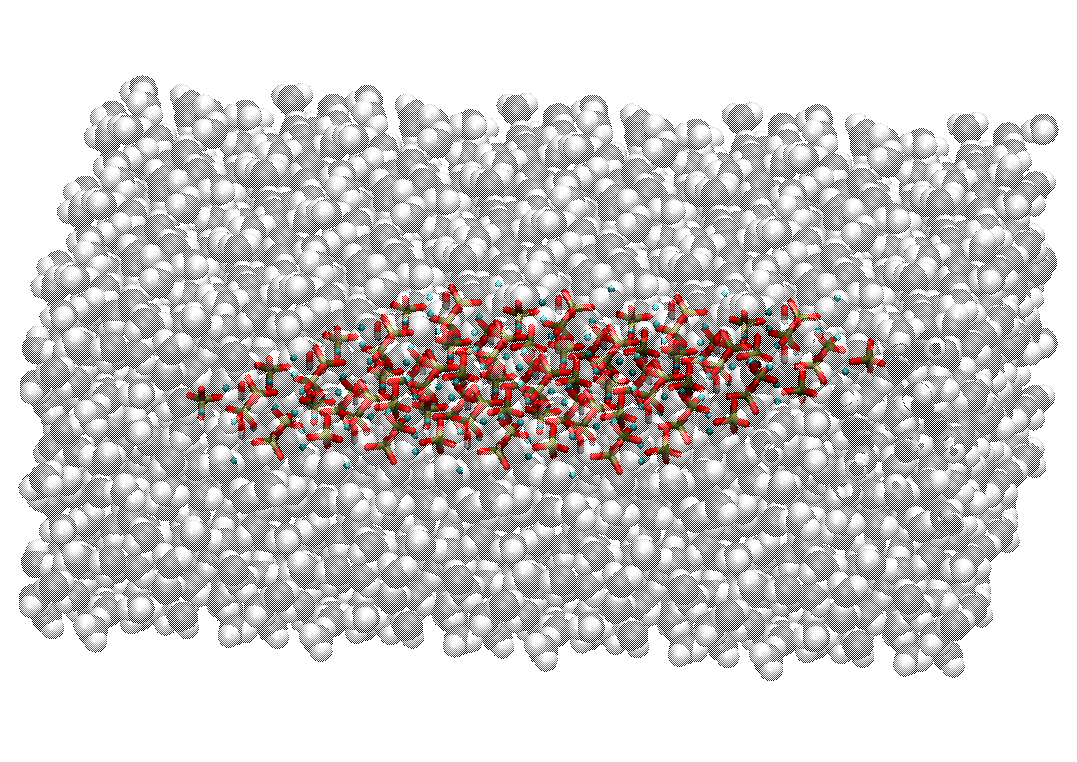
Click here to go to: Hydroxyapatite-Water Tutorial
Before we perform the first and second examples, we need to have the INTERFACE force field in the AMBER software package. The AMBER compatible INTERFACE force field files could be downloaded here: interface_v1_5.dat, fcc_metals.lib, and leaprc.interface_v1_5, which are referred from the INTERFACE force field version 1.5 released here: INTERFACE-MD. In order to prevent conflicts with the available AMBER force fields, the normal atom types in the INTERFACE have been named with pure numbers, the FCC metals have been named with 0 as suffix, while the atomic ions have been named using p to replace the "+" of the atom types in the original INTERFACE force field version 1.5. You can download the INTERFACE parm file and move it to $AMBERHOME/dat/leap/parm/ directory, download the FCC metal lib file and move it to $AMBERHOME/dat/leap/lib/ directory, and download the leaprc file and move it to $AMBERHOME/dat/leap/cmd/ directory, then you can use them as the normal AMBER files. Note that these AMBER compatible INTERFACE force field files are trial, which are not guaranteed to work, and the listed tutorials herein are just modest tested by the author while longer simulation may reflect their deficiencies. Please check these force field files or the prmtop and inpcrd files carefully before proceeding to the minimization and MD simulation. ParmEd could offer help for these checks (e.g. commands such as printBonds, printAngles, printDihedrals, printDetails).
References:
[1] Hendrik Heinz, Tzu-Jen Lin, Ratan K. Mishra, and Fateme S. Emami, "Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: The INTERFACE force field", Langmuir, 2013, 29, pp. 1754-1765