"The Jupyter Notebook is a web application that allows you to create and share documents that contain live code, equations, visualizations and explanatory text. Uses include: data cleaning and transformation, numerical simulation, statistical modeling, machine learning and much more."
pytraj is a Python package binding to the popular cpptraj program. pytraj is written do extend the flexibility of cpptraj and to expose cpptraj's functionality to Python's ecosystem, such as numpy, pandas, matplotlib, ...
$ amber.jupyter notebook
(or jupyter notebook if you did not allow AMBER to install Miniconda)
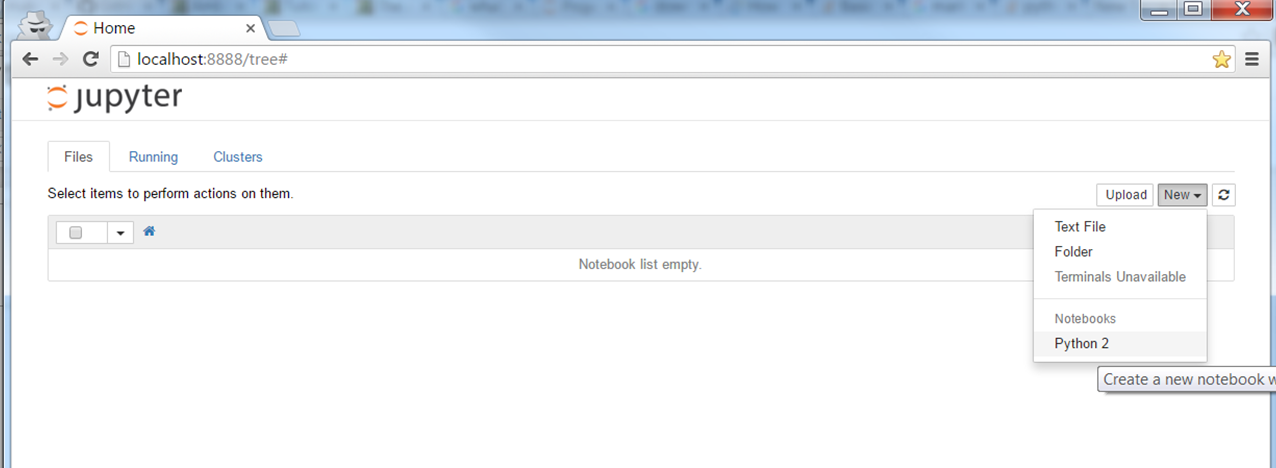
You should expect to see a blank directory (or with a list of files)
Choose New --> Python 2 (or Python 3, depending on which version you're using)
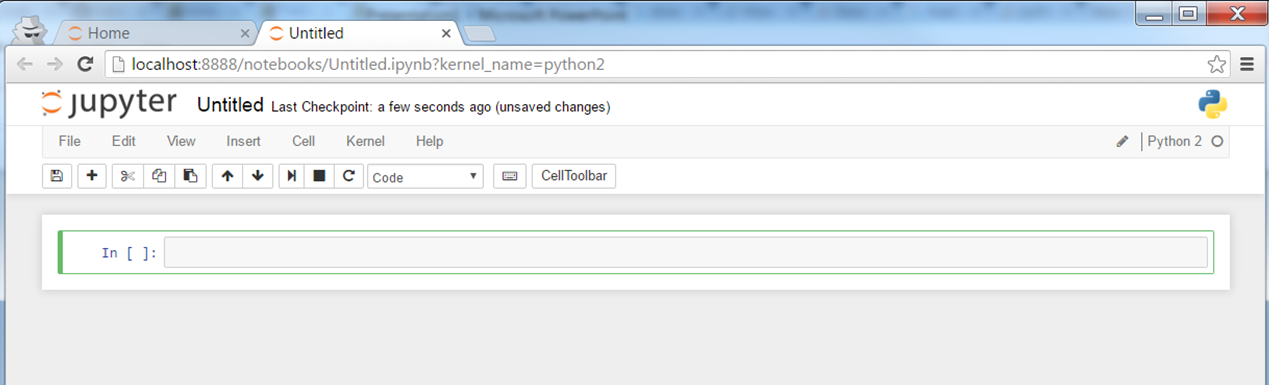
You should expect to see
How to follow this tutorial? Copy and paste each command in this tutorial to the notebook, hit "Shift-Enter" to run and to create new Cell. Check Tips in the end of this tutorial for useful commands.
If you want to use the notebook remotely (run it in your remote cluster and view it in your desktop), please check remote notebook tutorial
Note: This tutorial is written in the Jupyter notebook, so you can download and run it too.
from__future__importprint_functionimportwarningswarnings.filterwarnings("ignore",category=DeprecationWarning)# avoid warning message to make the notebook nicerimportpytrajaspttraj=pt.load('data/trpzip2.gb.nc',top='data/trpzip2.ff10.mbondi.parm7')
# compute pairwise RMSD for first 50 snapshots and skip every 10 framesmat=pt.pairwise_rmsd(traj,mask=":1-13&!@H*",frame_indices=range(0,500,10))print(mat)
In this case, we are computing phi and psi angle for residues 2 to 12. We tell pytraj to convert raw data to pandas's DataFrame
to better visualization in notebook