


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008
Building your own Custom Residues (old version)
Formerly known as TUTORIAL A1: Simulating a Solvated Protein that Contains Non-Standard Residues (Simple Version)
By Ross Walker
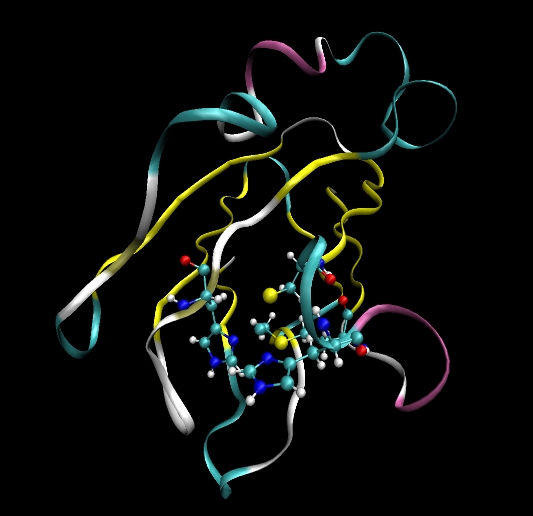
In tutorials 1 to 3 all the systems we studied contained only standard amino acid or nucleic acid residues. As such we did not need to create units for non-standard residues and provide our own parameters. In this tutorial we will look at one method for creating a non-standard residue. Note, there are many different ways to achieve the same thing in AMBER this is just one such way. Also, note, this tutorial provides an example for setting up a simple simulation of a protein containing a copper ion. Many assumptions are made here such as the fact that the copper can just be treated as having a +1 charge and that being bound to the copper does not change the parameters of surrounding residues. This is not strictly true but is useful for a first attempt. Ultimately, if serious simulations were to be done it would be necessary to create new units for the copper bound residues, re-fit all the charges for these regions and also provide new parameters for the bonds angles and dihedrals that are affected by the copper atom.
In this tutorial we will be aiming to setup a protein simulation of Plastocyanin in explicit solvent. In order to do this there are a number of things we will need to do:
-
Plastocyanin contains a copper ion (Cu) bound to four amino acids. His37, Cys84, His87 and Met92. We will need to modify these residues such that we can bind them to the copper ion. We will also need to supply parameters for these new bond types (and the corresponding angle and dihedral types these bonds create).
-
The PDB file for plastocyanin (1PLC) contains crystallographic waters which we should keep. However, only the oxygen positions are specified so we will use XLeap to add the missing protons. We will have to minimise these positions, however, before running production simulations.
-
Somewhat unusually the PDB file contains explicit protons for the protein. This is the same situation as tutorials 2 and 3, the proton naming convention used does not match IUPAC specifications. In this case we will correct this by removing the protons and letting Leap add them for us.
-
Using the most probable protonation states (at neutral pH) results in the Plastocyanin protein having an overall charge of -9. As such we will add 9 Na+ counterions around the system to neutralise it.
This will take a bit of work but it is significantly easier to do in AMBER 8 than it used to be.
This tutorial consists of four sections:
-
section1 : Do some editing of the PDB file.
-
section2 : Creating the non-standard CUA unit.
-
section3 : Loading the protein PDB and creating a Library File
-
section4 : Creating the Prmtop and Inpcrd Files
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2008