


Copyright (C) Pengfei Li & Kenneth M. Merz Jr. 2015
MCPB & MCPB.py -- Workflow
The general workflow for the parameterization a metal site using MCPB & MCPB.py can be summarized as shown in the following Figure:
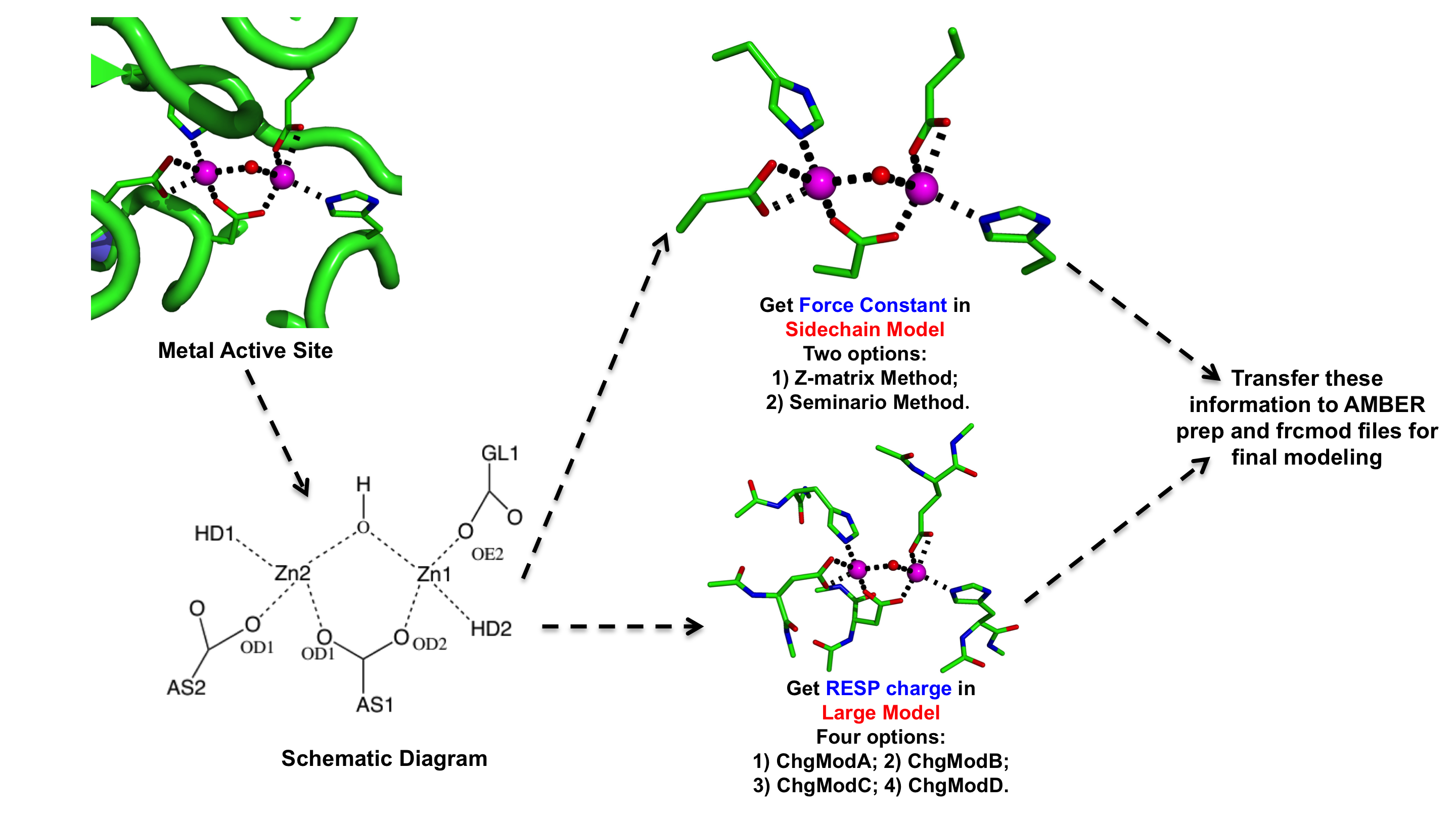
Normally the parameterization process consists of two parts:
1) Obtain the parameters for the bonded part (bond and angle), MCPB will neglect the dihedral part in the parameterization process since Metal-Ligand torsion barriers are below kT.
2) Obtain the point charge parameters for the electrostatic part of the potential.
To balance the speed and accuracy of the parameterization, the two parts of process are based on different metalloenzyme active site models:
1) The first part is based on the sidechain model, which only considers the sidechains of the ligating residues terminated with a methyl group. For this step, the user has two options to chose:
Z-matrix method (Both in MCPB and MCPB.py) The bond and angle force constants are determined from the Cartesian Hessian matrix Seminario Method (Both in MCPB and MCPB.py) The bond and angle force constants are determined from the sub matrices of the Cartesian Hessian matrix Empirical Method (Only in MCPB.py) The bond and angle force constants are determined from an empirical method developed by Li et al. in Merz Research Group, current version only supports bonded model of Zinc ion 2) The second step is based on the large model, in which the sidechains and their ACE and NME terminated backbones are included. For this step users have four options:
ChgModA Allow the charges of the all the atoms of the ligating residue to change ChgModB Restrains the charges of the backbone heavy atoms (CA, N, C, O) to the values in AMBER FF94 ChgModC Restrains the charges of all the backbone atoms (CA, H, HA, N, HN, C, O) to the values in AMBER FF94 ChgModD Restrains the charges of not only all the backbone atoms but also the sidechain CB atom to the values found in AMBER FF94
The Seminario/ChgModB combination gave the best results while Seminario/ChgModD also showed promising performance in the tests of the original paper, which is consistent with that the point charges of the backbone atoms play roles in the torison parameters fittings. [1]
Click here to go to: MCPB Example
Click here to go to: MCPB.py Example
Peters et al.[1] have developed zinc AMBER force field (ZAFF) for 4-coordinated zinc metal centers (not suitable for 5- or 6- coordinated zinc centers). Users who are going to model similar metal centers can use these parameters directly. Here is the link for the ZAFF modeling tutorial:
Click here to go to: ZAFF Moddeling Tutorial
References:
[1] Martin B. Peters, Yue Yang, Bing Wang, László Füsti-Molnár, Michael N. Weaver, and Kenneth M. Merz, Jr. "Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF)", J. Chem. Theory Comput., 2010, 6, pp. 2935-2947