


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018
Section 3: Running and analyzing production simulations at pH 7.0 and Redox Potential -203 mV
Running the simulations
The experimental standard Redox Potential of NAcMP8 with an imidazole molecule as the axial ligand has been measured as -203 mV vs NHE and at pH 7.0 [2]. This closely represents our system (NAcMP8 with a histidine peptide axially connected), shown in the figure at the Introduction. The reference energies for the HEH residue have been fitted to reproduce this result. Therefore, when simulating our system at pH 7.0 and redox potential -203 mV we must obtain a fraction of reduced species for the HEH residue close to 50%, if the simulation is converged.
We now present the input files of the production simulations at pH 7.0 and redox potential -203 mV in both implicit and explicit solvent. In this stage, the backbone restraints are not applied. For simplicity, in this tutorial we will perform shorter production simulations than the ones performed in our publication [1]. Here, simulations are 20 ns in implicit solvent and 40 ns in explicit solvent.
| prod.implicit.mdin | prod.explicit.mdin |
|
Production Stage in Implicit Solvent at pH 7.0 and redox potential -203 mV &cntrl icnstph=1, ! Switch for constant pH (=1, implicit solvent) ntcnstph=5, ! Protonation state change attempt every ntcnstph steps solvph=7.0, ! Solvent pH icnste=1, ! Switch for constant redox potential (=1, implicit solvent) ntcnste=5, ! Protonation state change attempt every ntcnste steps solve=-0.203, ! Solvent redox potential (in Volts) ntt=3, ! Temperature scaling (=3, Langevin dynamics) gamma_ln=10.0, ! Collision frequency of the Langevin dynamics in ps-1 ntc=2, ! SHAKE constraints (=2, hydrogen bond lengths constrained) ntf=2, ! Force evaluation (=2, hydrogen bond interactions omitted) ntb=0, ! Boundaries (=0, non-periodic) cut=1000.0, ! Cutoff dt=0.002, ! The time step in picoseconds nstlim=10000000, ! Number of MD steps to be performed ig=-1, ! Random seed (=-1, get a number from current date and time) ntwr=10000, ! Restart file written every ntwr steps ntwx=10000, ! Trajectory file written every ntwx steps ntpr=10000, ! The mdout and mdinfo files written every ntpr steps ioutfm=1, ! Trajectory file format (=1, Binary NetCDF) igb=2, ! GB model saltcon=0.1, ! Salt concentration irest=1, ! Flag to restart the simulation ntx=5, ! Initial condition (=5, coord. and veloc. read from the inpcrd file) / |
Production Stage in Explicit Solvent at pH 7.0 and redox potential -203 mV &cntrl icnstph=2, ! Switch for constant pH (=2, explicit solvent) ntcnstph=100, ! Protonation state change attempt every ntcnstph steps ntrelax=100, ! Number of relaxation steps after a successful protonation state change solvph=7.0, ! Solvent pH icnste=2, ! Switch for constant redox potential (=2, explicit solvent) ntcnste=100, ! Protonation state change attempt every ntcnste steps ntrelaxe=100, ! Number of relaxation steps after a successful redox state change solve=-0.203, ! Solvent redox potential (in Volts) ntt=3, ! Temperature scaling (=3, Langevin dynamics) gamma_ln=2.0, ! Collision frequency of the Langevin dynamics in ps-1 ntc=2, ! SHAKE constraints (=2, hydrogen bond lengths constrained) ntf=2, ! Force evaluation (=2, hydrogen bond interactions omitted) ntb=1, ! Boundaries (=1, constant volume) cut=8.0, ! Cutoff dt=0.002, ! The time step in picoseconds nstlim=20000000, ! Number of MD steps to be performed ig=-1, ! Random seed (=-1, get a number from current date and time) ntwr=10000, ! Restart file written every ntwr steps ntwx=10000, ! Trajectory file written every ntwx steps ntpr=10000, ! The mdout and mdinfo files written every ntpr steps ioutfm=1, ! Trajectory file format (=1, Binary NetCDF) iwrap=1, ! Translate water molecules into the original simulation box igb=0, ! GB model (=0, explicit solvent) saltcon=0.1, ! Salt concentration irest=1, ! Flag to restart the simulation ntx=5, ! Initial condition (=5, coord. and veloc. read from the inpcrd file) / |
|
For implicit solvent simulation, the command |
For explicit solvent simulation, the command |
As can be seen above, the meaning of each input variable is explained within our input files. As the input files indicate, we may perform protonation and redox attempts at different intervals, and also perform a different number of relaxation steps in explicit solvent for successful protonation or redox state changes. In our simulations, we perform protonation and redox state change attempts in the same MD steps because ntcnstph = ntcnste. In our code, when protonation and redox state change attempts happen in the same MD step, the protonation state change attempts are performed first. In our explicit solvent simulation, the number of relaxation steps is also the same for constant pH and for contant redox potential because ntrelax = ntrelaxe. However, in simulations where ntrelax is different from ntrelaxe, when protonation and redox state change attempts are to be performed in the same MD step the solvent relaxation is performed only once (if at least one state change is successful). Relaxation is performed after all state change attempts have been performed, using the biggest value between ntrelax and ntrelaxe as the number of relaxation steps.
Analysing the simulations
We will now analyze the constant pH and constant redox potential output files using cphstats and cestats, respectively. These tools have a very similar usage (a list of all available command-line flags can be seen using the --help flag). More information can be found in Amber's manual or here.
| Implicit Solvent: | Explicit Solvent: | |
|
Analyzing pH-active residues The following command cphstats -i mp8_is.cpin mp8_is.prod.cpout generates the following output: Solvent pH is 7.000 GL4 12 : Offset -2.538 Pred 4.462 Frac Prot 0.003 Transitions 864 PRN 15 : Offset -0.798 Pred 6.202 Frac Prot 0.137 Transitions 32916 PRN 16 : Offset -0.601 Pred 6.399 Frac Prot 0.200 Transitions 39006 Averag total molecular protonation: 0.341 Analyzing the redox-active residue The following command cestats -i mp8_is.cein mp8_is.prod.ceout generates the following output: Redox potential is -0.20300 V, temperature is 300.00 K HEH 14 : Offset -0.00080 V Pred Eo -0.20380 V Frac Redu 0.49228 Transitions 1455651 Average total molecular reduction: 2.49228 |
Analyzing pH-active residues The following command cphstats -i mp8_es.cpin mp8_es.prod.cpout generates the following output: Solvent pH is 7.000 GL4 12 : Offset -1.563 Pred 5.437 Frac Prot 0.027 Transitions 1338 PRN 15 : Offset -0.205 Pred 6.795 Frac Prot 0.384 Transitions 11290 PRN 16 : Offset -0.264 Pred 6.736 Frac Prot 0.353 Transitions 12062 Averag total molecular protonation: 0.763 Analyzing the redox-active residue The following command cestats -i mp8_es.cein mp8_es.prod.ceout generates the following output: Redox potential is -0.20300 V, temperature is 300.00 K HEH 14 : Offset 0.01595 V Pred Eo -0.18705 V Frac Redu 0.64954 Transitions 121339 Average total molecular reduction: 2.64954 |
As can be seen, the fraction of reduced species of the redox-active residue HEH in implicit solvent is close to 50%. For explicit solvent, this is not the case. This happens because we would need to run much longer to achieve convergence using C(pH,E)MD only, as we show in reference [1] (in the next section we will discuss how we can improve the convergence efficiency in our simulations).
More accurate Eo predictions than the one shown by the cestats output require simulations at different values of redox potential; similarly, simulations at different pH values are needed for proper pKa predictions. This will be done in the next section.
Visualizing the trajectories in VMD
Beginners may find introductory VMD instructions in the Basic Tutorial 2. We can use VMD to visualize C(pH,E)MD trajectories, and with the use of the vmd_cphemd.py script we can visualize in each trajectory frame the protonation state of each pH-active residue and the redox state of the redox-active residue. To represent each protonation state we move the dummy hydrogens that are not protonated 99 Angstroms away from its position (because of that, the use of Dynamic Bonds for the pH-active residues is needed for proper visualization). The different redox states are shown by different colors. Using the default options, the oxidized state corresponds to the red color and the reduced state to the blue color.
The procedure we will show below is the same for both implicit and explicit solvent simulations. However,
for simplicity, we will only show the procedure using the implicit solvent files.
The vmd_cphemd.py script can be executed with the following
command (see ./vmd_cphemd.py --help for full usage):
./vmd_cphemd.py -O -cpin mp8_is.cpin -cpout mp8_is.prod.cpout -cein mp8_is.cein -ceout
mp8_is.prod.ceout
This command generates the cphemd_script.tcl file.
cphemd_script.tcl is a TCL script we will enter
in VMD after loading the topology and trajectory files to obtain our desired outcome.
The vmd_cphemd.py script was generalized for C(pH,E)MD from a
script for CpHMD written by Jason Swails reported here.
The vmd_cphemd.py script can be used in simulations where only the constant pH or only the constant redox potential option is active. If the simulation was restarted, the CPOUT and CEOUT files must be listed in the -cpout and -ceout flags, respectively, in the same order as the trajectory files are loaded into VMD.
We now present instructions on how to apply the generated TCL script into VMD.
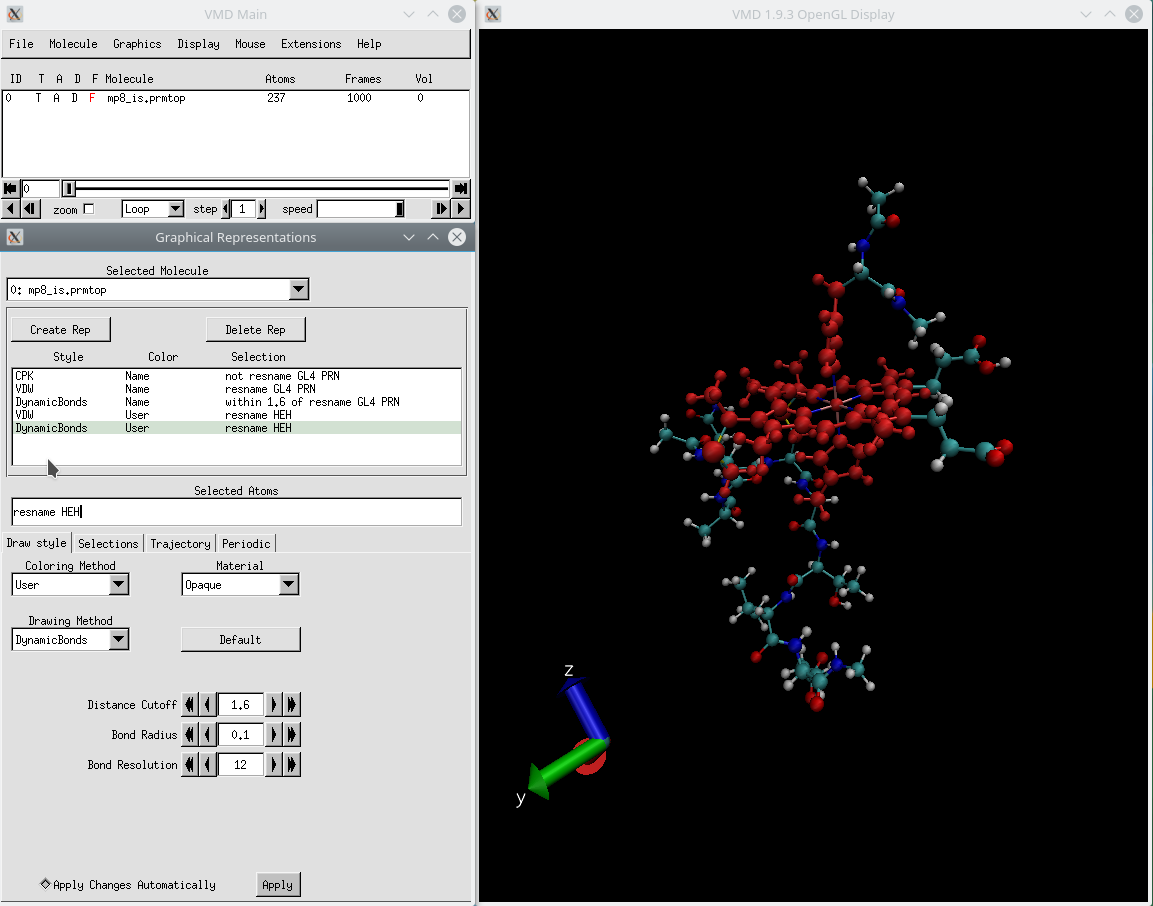
Operating VMD
To load the Amber input files, either:
- In your terminal, simply load VMD with the following command: vmd mp8_is.prmtop mp8_is.prod.nc
Or:
- Open VMD
- In the console titled VMD Main, click File, followed by New Molecule
- Load the topology file, mp8_is.prmtop, (AMBER7 Parm) format, followed by the trajectory file, mp8_is.prod.nc, (NetCDF AMBER,MMTK) format
Then, the following settings must be done (make sure to follow these instructions carefully):
- In VMD's terminal, type: source cphemd_script.tcl
- In the console titled VMD Main click Graphics, followed by Representations
- Change the Drawing Method in Graphical Representations console to CPK, and under Selected Atoms type: not resname GL4 PRN
- Click the tab Create Rep, change the Drawing Method to VDW, reduce the Sphere Scale to 0.3, and under Selected Atoms type: resname GL4 PRN
- Click the tab Create Rep, change the Drawing Method to Dynamic Bonds, reduce the Bond Radius to 0.1, and under Selected Atoms type: within 1.6 of resname GL4 PRN
- Create one new replica for each of the last two replicas created (VDW and DynamicBonds)
- On the last two replicas just created, change the Coloring Method to Trajectory→User→User, and under Selected Atoms type: resname HEH
- In the Graphical Representations window, go to the Trajectory tab, and make sure to set the Color Scale Data Range from 0.0 to 100.0
This is how your final screen in VMD should look like:
Click here to go back to Section 2
References
[1] Vinícius Wilian D. Cruzeiro, Marcos S. Amaral, and Adrian E. Roitberg, "Redox Potential Replica Exchange Molecular Dynamics at Constant pH in AMBER: Implementation and Validation", J. Chem. Phys., 149, 072338 (2018). DOI: https://doi.org/10.1063/1.5027379
[2] H.M. Marques, I. Cukrowski, and P.R. Vashi, "Co-ordination of weak field ligands by N-acetylmicroperoxidase-8 (NAcMP8), a ferric haempeptide from cytochrome c, and the influence of the axial ligand on the reduction potential of complexes of NAcMP8", J. Chem. Soc. Dalt. Trans., 8, 1335 (2000).
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018