


(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018
Constant pH and Redox Potential MD Example: Predicting pH-dependent Eo values
By Vinícius Wilian D. Cruzeiro
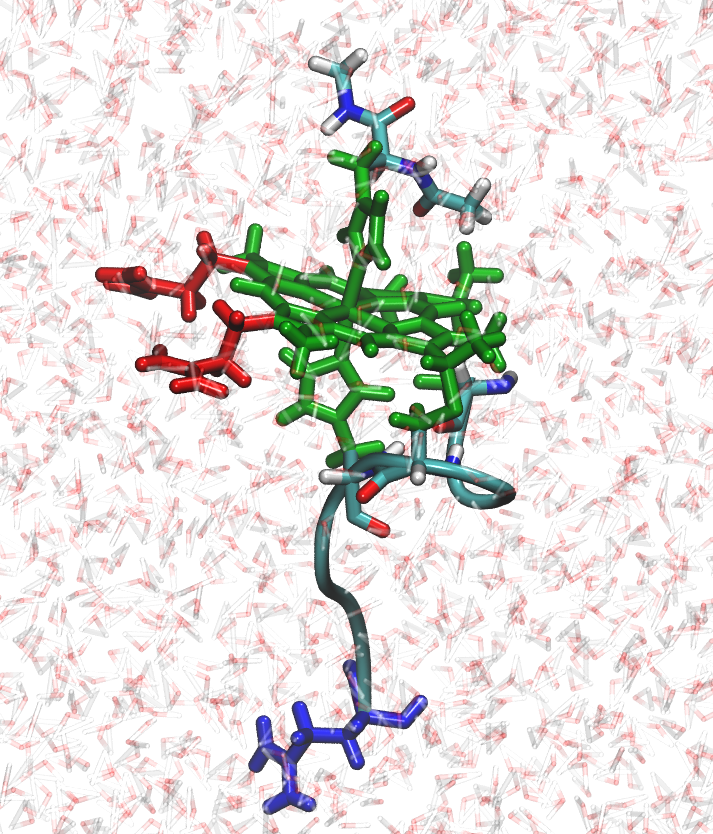
In this tutorial, we will learn how to use the software available in Amber and AmberTools to perform molecular dynamics simulations at constant pH and Redox Potential (C(pH,E)MD). The C(pH,E)MD method is available since Amber version 18. In this tutorial, AmberTools will be used to both prepare input files and to analyze results. The molecular dynamics simulations can be executed using either sander or pmemd. The C(pH,E)MD method can be executed in serial, parallel (sander.MPI or pmemd.MPI), and is also available using pmemd's GPU-accelerated code (pmemd.cuda), which provides great speedup in comparison to calculations that use CPUs only.
As many redox processes are also performed at a fixed value of pH, having an efficient computational method capable of describing experimental measurements done at both constant pH and redox potential is very important. The implementation of the C(pH,E)MD method in Amber is described in Ref. [1]. The process of creating input files and executing the simulations is somewhat similar to what is done in constant pH simulations. Therefore, for a better understanding of this tutorial, it is recommended that the reader has some familiarity with constant pH simulations (Amber's CpHMD tutorial can be found here).
This tutorial assumes familiarity with Ref. [1], and will outline only the practical steps for running C(pH,E)MD simulations. We will be covering simulations in both implicit and explicit solvent models. For a more detailed discussion about how enhanced sampling can be performed on C(pH,E)MD using replica exchange simulations, please refer to Ref. [2]. Different applications of this methodology have been presented and discussed on Ref. [3].
In this tutorial, we will be performing simulations for N-acetylmicroperoxidase-8 (NAcMP8) axially connected to a histidine peptide (see figure above), the same system used in Ref. [1]. NAcMP8 is a small peptide that contains a single heme group.
This tutorial consists of four sections:
1) Section 1: Preparing the initial structure and input files.
2) Section 2: Performing minimization, heating, and equilibration stages.
3) Section 3: Running and analyzing production simulations at pH 7.0 and Redox Potential -203 mV.
4) Section 4: Running and analyzing production simulations at different pH and Redox Potential values.
References
[1] Vinícius Wilian D. Cruzeiro, Marcos S. Amaral, and Adrian E. Roitberg, "Redox Potential Replica Exchange Molecular Dynamics at Constant pH in AMBER: Implementation and Validation", J. Chem. Phys., 149, 072338 (2018). DOI: https://doi.org/10.1063/1.5027379
[2] Vinícius Wilian D. Cruzeiro, and Adrian E. Roitberg, "Multidimensional Replica Exchange Simulations for Efficient Constant pH and Redox Potential Molecular Dynamics", J. Chem. Theory Comput., 15, 871–881 (2019). DOI: https://doi.org/10.1021/acs.jctc.8b00935
[3] Vinícius Wilian D. Cruzeiro, Gustavo T. Feliciano, and Adrian E. Roitberg, "Exploring Coupled Redox and pH Processes with a Force-Field-Based Approach: Applications to Five Different Systems", J. Am. Chem. Soc., 142, 3823–3835 (2020). DOI: https://doi.org/10.1021/jacs.9b11433
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Vinícius Cruzeiro. 2018