


Fundamentals of LEaP
By Pengfei Li and David Cerutti
Background Information
The LEaP program is a portal between many chemical structure
file types (.pdb and .mol2, primarily), and the Amber
model parameter file types such as .lib, .prepi,
parm.dat, and .frcmod). Each of the parameter files
contains pieces of information needed for constructing a simulation, whether
for energy minimization or molecular dynamics. Given a complete set of
parameters (the molecular model, which we call a "force field"),
LEaP will generate an AMBER topology file (generally, the file
extension is ".prmtop", although ".parm7" and
".top" are synonymous for the same file type) and coordinate file
(appropriate file extensions for the products of LEaP include
".inpcrd" and ".crd"). LEaP functions
within a larger workflow described in Section 1.1 of the
Amber Manual.
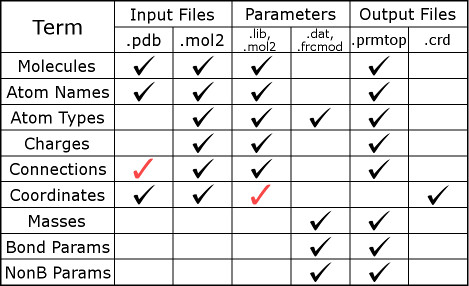
The figure below illustrates the information contained in different file
types. A black checkmark in the figure indicates that such files are
relevant to LEaP. A red checkmark in
the figure indicates the file contains additional information that may not be
relevant to LEaP. More detailed information about the contents of
some files and their formats can be found on the
AMBER file formats webpage, and
in Section 14.1 of the
AMBER 2016 Manual.
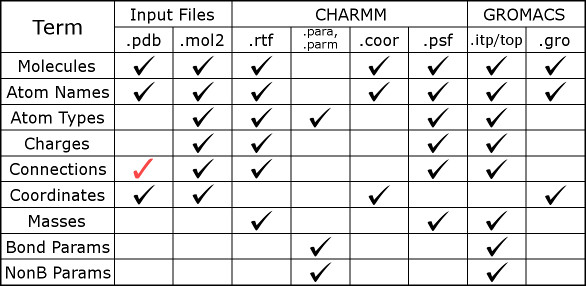
Here we also have a figure for modeling files in CHARMM and GROMACS.
How Does LEaP Work?
The LeAP program collates parameters in order to make a
complete description of a molecule. To see this in action, just read through
one of the leaprc files in $AMBERHOME/dat/leap/cmd/.
Each of them is read as a script: there are declarations of atom types and
residues, as well as calls to load files containing parameters and residue
definitions. For instance, our recommended protein force field is encoded in
leaprc.protein.ff14SB, a file you'll be calling often. In that
leaprc, you will find a call to load parm10.dat (a
"full" force field parameter file) as well as frcmod.ff14SB (an
auxiliary parameter file which has FORce field MODifications).
In this case, frcmod.ff14SB supplements parm10.dat,
but since it is specified later any conflicts will favor the
frcmod. An alternative, equally modern protein force field can be
found in leaprc.protein.ff15ipq, and this file calls only the main
parameter file because all of its parameters were derived at the same time.
In all leaprc files, there must also be libraries
(lib files). It's one thing to have parameters, but it won't be
of much help until the units of our molecule are clearly defined in terms of
those parameters. As is indicated in the precedning chart, the residue
libraries contain some of their own parameters: the charges. It may seem like
an odd division of the material, but it owes to the way the Amber force fields
work. There are alternative paradigms for defining a force field, which are
well beyond the scope of this tutorial, but in short applying them would
require a different program.
Once LEaP knows all of the parameters, it tries to completely
define all aspects of a structure it has been given. There is some degree of
fault tolerance: LEaP can be presented with incomplete residues,
or requests to simply build residues, and it will comply so long as it knows
what the residues are made of. However, if LEaP does not know
what a residue is, or more often it encounters atom names inside a residue that
do not quite match its libraries, the program will throw an error message and
you will not get a system ready for simulations until that's fixed. The
coordinates that come out of LEaP reflect the structure it was
given, after filling out any incomplete residues or constructing what it was
told to make. The topology that comes out of LEaP is a complete
description of how that system will behave. The simulation is needed because
the system is almost certainly so complex that analytic solutions are not to
be found.
| Structure File | Library Files | Parameter Files | Topology | Coordinates | |
|
Molecules Atom Names Connections Coordinates |
Units (Residues) Atom Names Atom Types Charges Connections Coordinates Masses Bonded Params Nonbond Params |
Atom Types Masses Bonded Params Nonbond Params |
→ → → |
Units (Residues) Atom Names Atom Types Charges Connections Masses Bonded Params Nonbond Params |
Coordinates |
"Standard" residues: AMBER has library files for standard amino
acids, nucleic acids, lipids, and sugars. These libraries are designed to
work with established parameter sets, as defined in their respective
leaprc files. In Amber16 and later versions, we have defined
leaprc files for different descriptions of proteins, nucleic
acids, and solvents (including a number of water models), so that you can mix
and match as you need. You include these leaprc files in your
LEaP input by simply stating
"source leaprc.protein.ff14SB" and so forth; can't find the files?
That's because they're stashed away in $AMBERHOME/dat/leap/cmd/,
but LEaP knows where to look in order to find the standard
includables much like the C compiler gets run with a default directory for its
standard libraries. Bear in mind that not all combinations of the models we
provide are recommended, but also that we went to the trouble of making these
files because we think that individually they are good options.
Non-standard residues: Library files may exist for nonstandard
residues (for instance, you may find them on the web), but you should always
check their contents and the means by which they were derived. In most cases
users will simply need to created a corresponding .lib file for
themselves. This process takes the antechamber program as
described in a
separate tutorial.
The antechamber program will generate its own .frcmod
files for custom-built residues, typically using the
General Amber Force
Field, but the exact source of parameters is subject to some user control.
The .prepi files that antechamber produces easily can
be read by LEaP to create library files proper, or just treated as
library files themselves. In most cases antechamber will also
create a .frcmod file containing custom parameters, which is also
then needed by LEaP when building the system.
The pdb4amber program: While we make our best effort to
provide force fields that plug into the structures the PDB and other databases
hold, there are multiple atom naming conventions and other aspects of
structures that prevent us from being universally compatible. To mitigate this
problem, we offer the pdb4amber program which modifies atom and
residue names into conventions that the rest of our programs will understand.
It's mostly for proteins, but it can be applied to other biomolecules. There
are some limitations to the program, such as a limit of 100,000 atoms owing to
the PDB CONECT format. The program will provide a list of options
if run with no arguments; a typical application is shown in the example below.
Adding bonds: One of the most common mistakes that can be made in
structure preparation is the failure to bond cysteine disulfide bridges, but
there are other connections to be scrupulous about. If a .pdb or
.mol2 structure file contains connectivity information that forms
a bridge between two cysteines, things will be all right. However, if only the
coordinates are taken from the file (perhaps you grep for "ATOM"
to eliminate crystallographic waters), the connections would need to be added
manually. If the disulfide bridge is lost, the CYS residues will not be
correctly converted to CYX and instead reduced (hydrogen added to the thiol) in
the simulation. Any bonds can be added to a structure in LEaP
input with the command:
bond <unit>.<residue #>.<atom name>
<unit>.<residue #>.<atom name>
The AMBER Manual
gives a more general description of how to add bonds, but this is the format
you will encounter most often. Make sure to relabel any cysteines that need
participate in disulfide bridges as CYX, not CYS as they appear in the Protein
Data Bank (PDB). The specifications of each atom are not
ambmask strings, and it is important to note that the residue
numbers you must provide are the positions of residues that appear in
the structure file, not the residue numbers that might be given in that
file. This makes a difference if you have a .pdb file that starts
its residue numbering at something relevant to biochemists, like CYS 14,
THR 15, GLU 16, CYS 17, … In that case, a disulfide bond between the
(relabeled) CYX 14 and CYX 17 of this structure (read from its parent
.pdb file as MyUnit) would be specified by: bond MyUnit.1.SG
MyUnit.4.SG.
The leap.log file: The most important output from leap will be piped
to the terminal, or whatever file you redirect it to (i.e. leap -f
InputFile.txt > OutputFile.txt), but there's another source of
output that will be constantly appended with verbose output from every
LEaP session run in the directory: leap.log. Clear
this file before running LEaP if you want to focus on the details
of what you are doing. Reading through leap.log, you will find
every command that you placed in your input file, and much more. It will
really show you how the leaprc files you reference are taken as
scripts: in fact, virtually every part of their contents could be specified
directly in the LEaP input file if it weren't alreayd written down
for you to include.
Error Messages: Even advanced AMBER users will make mistakes, but
fortunately LEaP reports most issues that it encountered. The key
is for all users to read the LEaP output carefully as the
errors are often easily overlooked. It is far better to spend a minute
checking the output only to find that nothing is wrong than to start a
simulation, run it for two weeks, and then review the outcome only to find that
the system had a subtle problem at the very start.
An Example with LEaP
This simple exercise in creating a solvated protein in water will apply the
principles we have just learned. We will use the snake venom toxin fasciculin,
1FSC.pdb, the ff14SB force
field to describe the protein, and the SPC/E water model. Because fasciculin
contains four disulfide bridges, we must rename the CYS residues participating
in such interactions (all of them, it turns out) to CYX. The CONECT records in
1FSC.pdb will take care of the bonds and CYS renaming when we apply
pdb4amber. The output of that operation is
here.
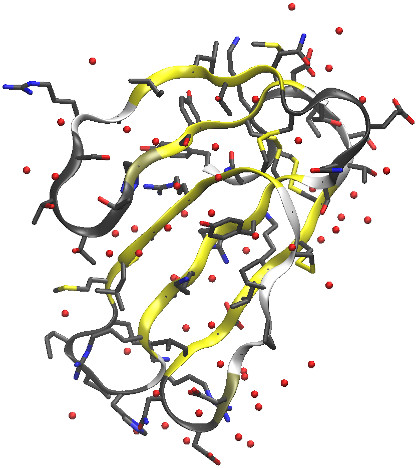
There are two atoms, marked "UNL" in the structure file, which we will
simply have to do away with--they are not part of the protein or any ligand,
and are probably just a problem for the crystallographic refinement. The final
.pdb file, before we apply tleap, is
here. The protein and all other
crystallographic waters can be seen in the figure below.
In order to solvate the system, we will make use of the
solvateOct command to put this thing in a regular, truncated
octahedral box. This is the closest thing to a sphere that Amber has, and the
most efficient way in terms of space (and thus particle count) to surround a
biomolecule with water. In the early days of MD we often did "shoe boxes" with
dimensions dictated by the shape and orientation of the biomolecule. The
trouble with this approach comes when simulating something that can tumble, and
the timescales our simulations routinely reach nowadays will pretty much
guarantee that will happen. In our simulation there will be periodic images of
the protein arrayed in an egg-crate layout (that's how the truncated
octahedron stacks), and we do not want a situation where the thing could touch
an image of itself. The settings below would be pretty good for running a
simulation with a 9 or 10Å cutoff; a buffer of 14Å is used, but the
water in that buffer region is going to settle somewhat around the protein so
the box volume will shrink a bit in the first 100-200ps of dynamics after a
barostat comes into play.
Another consideration: the fasciculin is charged, and the best practice is
to have a system that is overall neutral. To compensate for the +4 charge, we
will add four chloride ions with the addIons command. The
complete LEaP input is here:
source leaprc.protein.ff14SB
source leaprc.water.spce
fasciculin = loadPdb "1fsc_amb.pdb"
solvateOct fasciculin SPCBOX 14.0
addIons fasciculin Cl- 4
saveAmberParm fasciculin solvated_1fsc.top solvated_1fsc.crd
quit
The output from this exercise can be found
here, and the leap.log file is
here. The resulting
topology and
coordinates can now be fed into the
initial minimization for a typical MD simulation. Here is the system we
made, showing the octahedral shape of the box (this is looking head-on at one
of the square faces, even though that may not be obvious from the outline of
the water):

There is a tutorial about using the AMBER force field in CHARMM. In this way users can take advantage of the functions provided by the CHARMM software package. Interested users can check this link.