


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
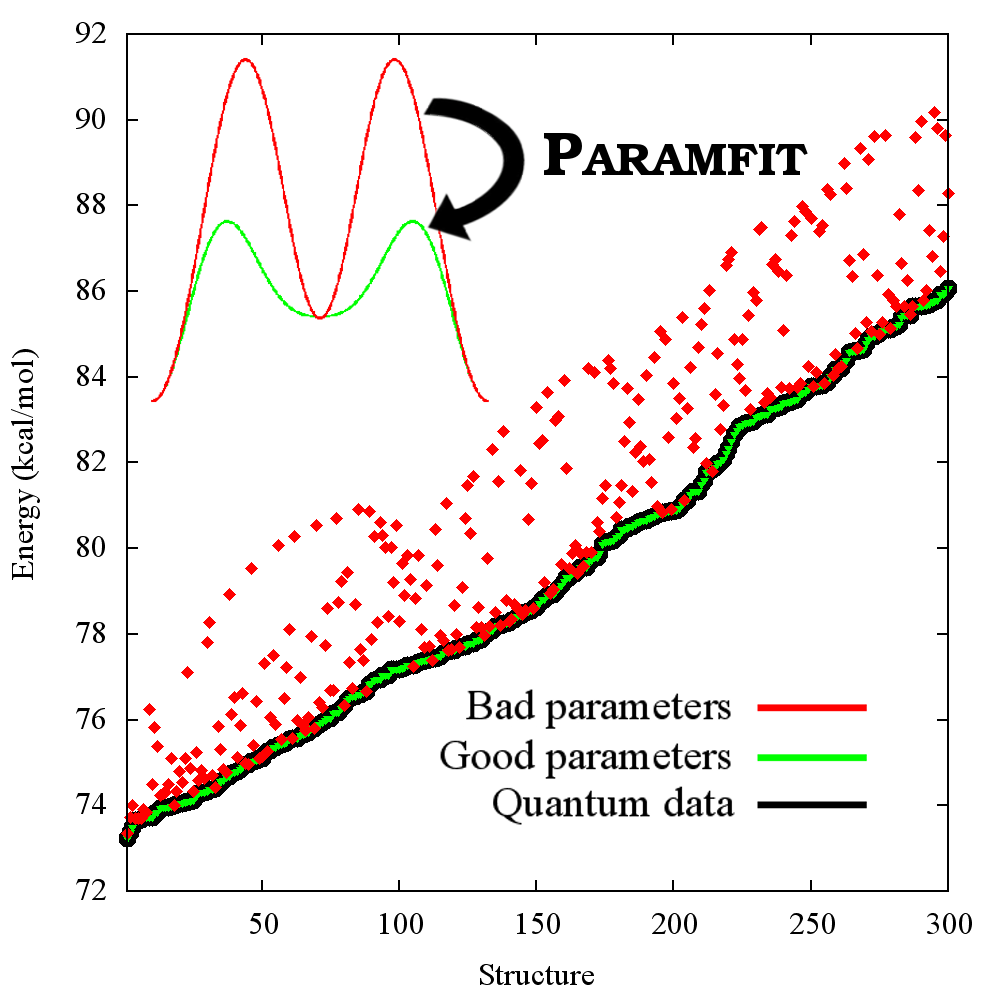
QUICK REFERENCE FOR RETURNING USERS
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
Introduction
This tutorial will guide you through deriving bonded parameters for a small molecule using Paramfit.AMBER includes a variety of force fields that will do a good job of describing atom interactions for the majority of systems. However, sometimes it is necessary to improve on these parameters. For some systems, parameters fail to adequately characterize interactions, or are simply not available.
Paramfit is a program available since AmberTools12 that allows for the generation or improvement of bonded force field parameters.
In this tutorial we will derive the potential for protein backbone dihedrals by fitting to several small polypeptides using Paramfit's support for weighted multiple molecule fits. This tutorial is also easily applicable to bonded parameter generation for a individual molecules.
Prerequisites
You will need AmberTools14 and a quantum package. This tutorial will use Gaussian, but there are free alternatives such as GAMESS, CP2K or MOPAC.
This tutorial assumes you have already assigned atom types, conducted a charge
derivation for the molecule using methods such as RESP or R.E.D, and that the nonbonded
parameters (Van der Waals, etc) have also been derived.
The tutorial also assumes familiarity with system setup in AMBER using xleap.
This tutorial will derive the amino acid backbone dihedrals, φ and ψ, from
ab-initio quantum data, as labelled on the following small peptide:
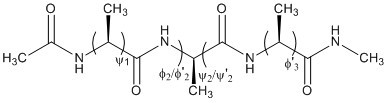
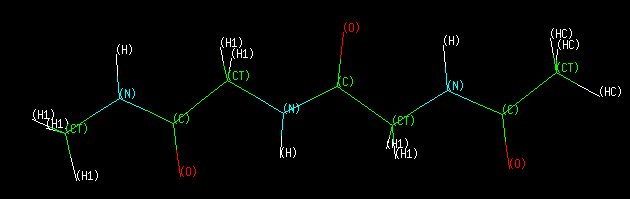
We will generate backbone parameters for this molecule, then conduct a multiple molecule
fit to derive the backbone parameter across all of them.
With AMBER ff99 atom types, these dihedrals are φ=C-N-CT-C and ψ=N-C-CT-N.
The general invocation of Paramfit is for a single molecule fit is as follows. You can click on each argument to be linked to the part of the tutorial where it is generated.
paramfit -i <Job Control File> \ -p <Topology File> \ -c <Coordinate File> \ -q <Quantum File>
WARNING: This example is somewhat contrived for the purposes of having a small system for this tutorial. You will most likely build your system in another fashion. However, loading it into leap will be done in the same way.