


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
Define parameters to fit
Browse through the output file from the K fitting run and notice how many parameters Paramfit lists as being found in the topology file. We do not want to re-derive all of these parameters (in fact, it would be very difficult to do so accurately without an enormous number of structures), so we will specify which parameters are to be fit.
Paramfit reads in the parameters to be fit from a "parameter file" that simply lists the bonds, angles, and dihedrals that should be adjusted in the fitting run. There is no initial value specified for these parameters- the file just specifies a binary of whether or not to fit each possible parameter.
For multiple structure fits, the same parameters must be present in each topology file, and only one parameter file is necessary to specify which ones are to be fit.
To create this file, we will use the SET_PARAMS run mode of Paramfit. Since we will do this only once, we will follow the wizard prompts to enter this mode without saving a job control file. We will be setting parameters using molecule A's topology, but it does not matter which molecule we use as the parameters of interest are present in all three molecules.
Now run Paramfit using the molecule A topology file and no job control file so that the wizard will start.
paramfit -p a.prmtop
Follow the prompts to set parameters, and specify a filename of prms.in. Then, Paramfit will prompt you if you want to fit each parameter in the topology file. For this tutorial, we want to fit all terms KP of the C-N-CT-C and N-C-CT-N dihedrals. (These dihedrals may appear on the list as CT-C-N-CT and N-CT-C-N since directionality is irrelevant). Specify you do not want to fit bonds or angles, you do want to fit dihedrals KP, and say "y" for all 4 terms of the two dihedrals.
Output: (Yours should be identical)
set_params.out
prms.in
Visualise structure quality
Remember that when we wrote the quantum input files, we received a lot of warnings about inadequate dihedral sampling. Now that we have defined which parameters are to be fit, we can selectively visualise the sampling of those dihedrals to evaluate whether or not our final parameters could be affected. This is a highly subjective process, and it is always better to just add more structures.
Create the following job control file:
job_scatterplots.in# Do a dummy fit RUNTYPE=FIT ALGORITHM=NONE NSTRUCTURES=20 COORDINATE_FORMAT=TRAJECTORY # Load the paramter file PARAMETERS_TO_FIT=LOAD PARAMETER_FILE_NAME=prms.in # Output dihedral scatterplots SCATTERPLOTS=YES
Run Paramfit without a job control file to enter the wizard, but provide the topology and coordinate files:
paramfit -i job_scatterplots.in -p a.prmtop -c A_valid_structures.mdcrd \
-q quantum_A.dat > scatterplots.out
Output: (Yours will differ)
scatterplots.out
16.diheq
21.diheq
The two .diheq files contain the phase found for each fitted dihedral type over all the structures. There may be more lines than structures in these files since a structure can contain multiple instances of a dihedral type, however in this case there is only one instance per file.
You can visualise these data using the scatterplots.sh script and Gnuplot. Run this in the same directory as the .diheq files and a visualization will appear:
$AMBERHOME/AmberTools/src/paramfit/scripts/scatterplots.sh
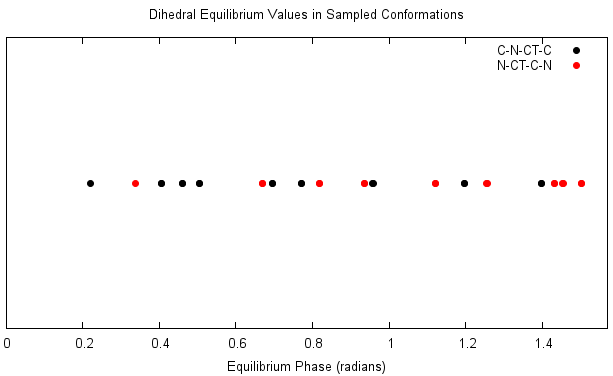 This looks like a reasonable spread for the C-N-CT-C dihedral, but the N-CT-C-N dihedral seems to be biased towards
larger phases and lacks samples below 0.5 radians. Let's create the job control file for the fit and run it anyway,
and see how this affects the resulting parameters.
This looks like a reasonable spread for the C-N-CT-C dihedral, but the N-CT-C-N dihedral seems to be biased towards
larger phases and lacks samples below 0.5 radians. Let's create the job control file for the fit and run it anyway,
and see how this affects the resulting parameters.