


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
System setup
The first step is to create a topology file containing the parameters to be fit. For our system, the dihedrals of interest are defined to contain only two terms, as found in parm99.dat:
N -CT-C -N 1 1.700 180.000 -1. N -CT-C -N 1 2.000 180.000 2. C -N -CT-C 1 0.850 180.000 -2. C -N -CT-C 1 0.800 0.000 1.
However, we want to regenerate the ff99SB correction, which features 4 terms for each of these dihedrals. A frcmod, or force field modification, file needs to be created containing all parameters that are to be fit. A reference for this file format may be found here.
NOTE: If you are defining any new atom types, all parameters involving that atom need to be defined in the frcmod file in order for leap to be able to save the topology file correctly. You may search for an initial guess for these parameters in other leap data files, or just set them to zero- Paramfit does not require an initial guess. You will also need to provide the MASS and NONB sections for this type per the frcmod format.
The parameters in this file may be set to an initial guess based on values found in other force fields, or may be simply set to zero or one- again, Paramfit does not require an initial guess. In this case, we will use the ff99 data and set the additional dihedral terms to zero.
dummy_parameters.frcmoddummy parameter file DIHEDRAL C -N -CT-C 1 0.000 0.000 -4. C -N -CT-C 1 0.000 0.000 -3. C -N -CT-C 1 2.000 0.000 -2. C -N -CT-C 1 1.700 0.000 1. N -CT-C -N 1 0.000 0.000 -4. N -CT-C -N 1 0.000 180.000 -3. N -CT-C -N 1 0.850 180.000 -2. N -CT-C -N 1 0.800 180.000 1.
Open xleap, and load or create the system with initial parameters. In this case, we will load the ff99 parameters and generate three molecules: alanine tripeptide, glycine tripeptide, and a tripeptide with both an alanine and a glycine, henceforth referred to as molecules a, b, and c.
source oldff/leaprc.ff99
a=sequence {ACE ALA ALA NME}
NOTE: If you defined any new atom types, you will need to specify that type to leap, including its element and hybridization. For example, if I was adding a type named "oc" that is a sp3 oxygen, I would add to the leap input the following: addatomtypes { { "oc" "O" "sp3" } }. See the AmberTools manual for more information on this command.
Then, define any parameters that leap by reading in the frcmod file you created, and save a topology and initial coordinates file for each molecule.
loadamberparams dummy_parameters.frcmod saveamberparm a a.prmtop a.inpcrd
WARNING: If there are any errors saving the topology, ensure that all missing parameters are defined in the frcmod file. The check command can assist with this.
Generating conformations
The next step is to generate a variety of molecular structures that sample conformation space involving the parameter to be fit. The simplest way to do this is to generate many random conformations, and discard any with unreasonably high energy (representing steric clash or overlap). We will use a script and leap to generate the random conformations.
View the molecules in xleap and select Display->Names. Note the atom names and residues involved in the parameters of interest.
Modify the gen_conformers.sh script to reflect the correct atom names and residues for your molecule. The script currently includes examples for generating random values for a bond, angle, and dihedral in molecule a.
This script creates a random structure set using leap and saves each structure individually in a created directory. Then, it uses cpptraj to collate the structures together into a coordinate file. The topology files written or the prmtop you saved in the previous step should be identical, so only one need be retained. A list of all the random values is saved in values.dat for reference.
./gen_conformers.sh
Output: (Yours may differ)
A_all_structures.mdcrd
Remove unreasonable conformations
If you visualize the resulting conformations, you will see that many of them
represent physically unreasonable structures. Running a fit on these
conformations could affect the parameters' accuracy for reasonable
structures, and would result in unnecessary quantum calculation.
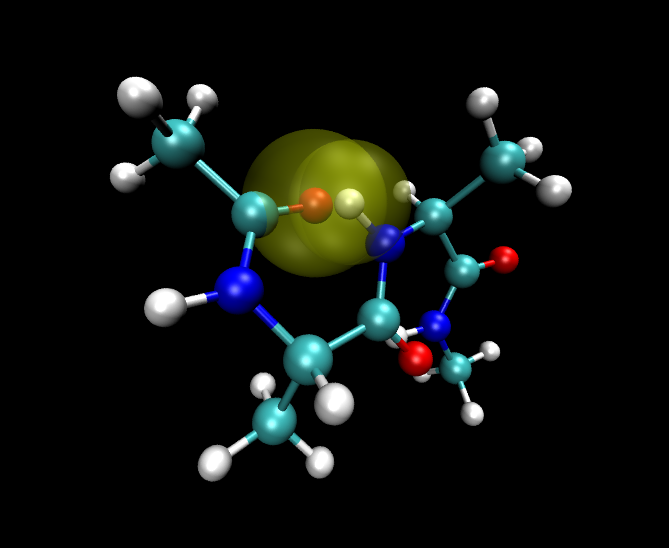 One unreasonable conformation for molecule A is shown here. The Van der Waals
radius of clashing atoms is highlighted.
One unreasonable conformation for molecule A is shown here. The Van der Waals
radius of clashing atoms is highlighted.
We will remove any structures with unreasonably high energy. An accurate energy calculation isn't necessary at this point; we just want to identify any insane structures, so the dummy or guess parameters in the frcmod file are adequate.
The prune_conformers.sh script evaluates the energy of each structure with Paramfit using whatever parameters are in the initial topology file, and if it is under 2000 kcal/mol (a high cutoff) it is added to the list of valid structures. Cpptraj then regenerates a coordinate file containing only those structures.
When running the script, record the number of structures that are valid. You will need this number later when creating quantum input files.
./prune_conformers.sh
Output: (Yours may differ)
A_valid_structures.mdcrd
Wrote 190 structures to A_valid_structures.mdcrd
Evaluate structure set quality
Now we have obtained a reasonable set of structures that randomly sample parameter space, but we have no guarantee that this sampling is adequate or provides good coverage of this space. For example, all the structures could represent fairly high energy conformations that would rarely be seen in molecular dynamics simulations. Paramfit fits to the input structure set, so the resulting set of parameters would describe those conformations very well but be undefined when calculating forces during a real simulation.
Fortunately, Paramfit provides functionality for verifying the quality of a parameter set. It has built in bounds checking features that will warn the user if:
- Derived bond equilibrium distance Req is more than 0.1Å from any length of that bond in input structures.
- Derived angle equilibrium value Θeq is more than π/20 radians from any value of that angle in input structures.
- A dihedral to be fit does not have samples every π/10 radians in phase in the input structure set.
The strictness of these checks may be adjusted by advanced users by changing BOND_LIMIT, ANGLE_LIMIT, and DIHEDRAL_SPAN in the job control file. Refer to the Paramfit section of the AmberTools manual for details.
NOTE: Do not change these values just to remove a warning! The bounds check warnings are meaningful and indicate problems with your input structures that can and will lead to inaccurate output parameters if not carefully validated!
Warnings for derived parameter values (bond and angle equilibrium distance) will appear following a fit, and will look something like:
WARNING: CT-CT bond has no sample structures larger than it, nearest sample is 0.7140 A smaller WARNING: CT-C bond has no sample structures within 0.1 A, nearest are 0.1032 and 0.2841 A WARNING: CT-CT-HC angle has no sample structures within 0.157 radians, nearest are 0.2001 and 0.1569 radians away
Seeing this error means that you need to carefully evaluate the output parameters to ensure that they are reasonable. The easiest way to do this is to provide additional input structures that have an equilibrium bond or angle value near the predicted equlibrium, and seeing if the fit still converges to the same result.
Dihedrals are checked at the beginning of the program, and will produce output like:
WARNING: C -N -CT-C dihedral is missing 4 data points in the range 1.8850 to 3.1416 radians.
WARNING: N -CT-C -N dihedral is missing 5 data points in the range 1.8850 to 3.1416 radians.
WARNING: Insufficient dihedral information in sample structures.
Your settings require at least 10 samples with data
at least every 0.314 radians (18.00 degrees).
Either 1) Add the missing input data or
2) Set DIHEDRAL_SPAN to a smaller value or
3) Set BOUNDS_CHECK to warn (not recommended).
Please read the help and/or documentation.
Since we will be fitting two dihedral terms, we care about the results of the dihedral span check. This check will be run at the beginning of the next step, when we create quantum input files.