


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
Multiple molecule fits
Now we have successfully derived parameters for a single molecule. This could be useful if you are missing parameters for a specific system, but what if you want more broadly applicable parameters that can describe a whole class of molecules? Paramfit addresses this force field development problem with its support for multiple molecule fits.
The fitting process is the same for single or multiple molecules, however the fitness equation is now summed over all molecules according to the following equation:
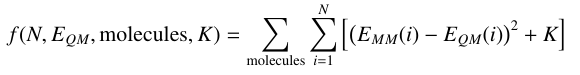
A few considerations: K must be derived separately for each molecule, and all parameters that are to be fit should be defined in each molecule. Paramfit will check for both of these things. Structure weighting is supported exactly as for single molecule fits- simply add an additional column to the quantum energy file that specifies the weights.
Paramfit is invoked for multiple molecule fits as follows:
paramfit -i <job control> -pf <prmtop list> -cf <mdcrd list>The syntax for the prmtop and mdcrd lists is discussed later.
Prepare two additional molecules for fitting, referring to previous sections of this tutorial for detailed instructions. Output files of these steps are available to download to check your work, but keep in mind your structure set and quantum energies will vary due to the random nature of the structue generation process.
This step is good practice for preparing systems for Paramfit. While you prepare the additional systems, consider the following:
- What should I modify in the gen_conformers.sh script?
- What do I need to change in the job control file, if anything?
- Do I need to re-generate the parameter file?
- Is this set of structures of good quality?
- Do I need to weight or ignore any of the structures?
Prepare additional molecules
For this tutorial, we will fit the φ and ψ amino acid backbone dihedrals across three small molecules, the first of which will be molecule A that was used for the single molecule tutorial.
Create two more topologies according to the directions in Part 1,
where molecule B is ACE-GLY-GLY-GLY-NME and molecule C is ACE-ALA-GLY-NME.
Molecule B topology
Molecule C topology
Generate conformations and prune unreasonable conformers as before
.
Molecule B valid structures
Molecule C valid structures
Similarly, conduct quantum calculations for both structures and process
the quantum output files, using the first 20 structures of each valid conformation set.
B quantum data file
C quantum data file
Now, derive K for both systems.
B fit K output
C fit K output
Do a sample fit to assess if any input structures need to be weighted, and
re-derive K for those systems. You will find that structure 8 of topology C
has unreasonably high energy and should be ignored.
weighted C quantum data file
C re-fit K output
Now you are ready to conduct a multiple molecule fit.
Define the prmtop list
The list of topologies, or "prmtop list," is a space delimited file, where each line contains the location of a topology file and the value of K for that file that we calculated earlier.
prmtops_list
a.prmtop -466382.135709 b.prmtop -416993.330876 c.prmtop -441694.818593
Create mdcrd list
The list of coordinate files, or "mdcrd list", is a space delimited file, where each line contains the location of a coordinate file, the number of structures to read from the file, and the location of the QM data file for those structures.
NOTE: These files should be in the same order as the prmtop list so that the topology on line N of the prmtop list corresponds to the coordinates on line N of the mdcrd list. Create the following file, remembering that molecules A and C have some high energy structures that are removed with weighting:
mdcrds_list
A_valid_structures.mdcrd 20 quantum_A_weighted.dat B_valid_structures.mdcrd 20 quantum_B.dat C_valid_structures.mdcrd 20 quantum_C_weighted.dat
Create job control file
By now, you should be familiar with using the wizard to create job control files. Generate a job control file for the multiple molecule fit using the standard AMBER energy equation and the genetic algorithm with the same settings as the single molecule fit. You can use the prms.in parameter file from the single molecule tutorial, as these backbone parameters are common to all three molecules.
Set the energy filename to multi_energy.dat.
Also specify a filename for a force field modification file, which is the easiest way to export parameters
from fits into other programs.
Download job control file
Conduct the fit
Now we can conduct the multiple molecule fit. Invoke Paramfit with the topology and coordinate lists. This will take longer than the single molecule fit as there are more structures.
paramfit -i job_multi_fit.in -pf prmtops_list -cf mdcrds_list \ > multi_fit.out
Check result quality
The resulting fit has a good R2 value. Output energy profiles will be written to filenames
specified at the end of the output. Plotting these with the provided script shows improvement for all three
structures.
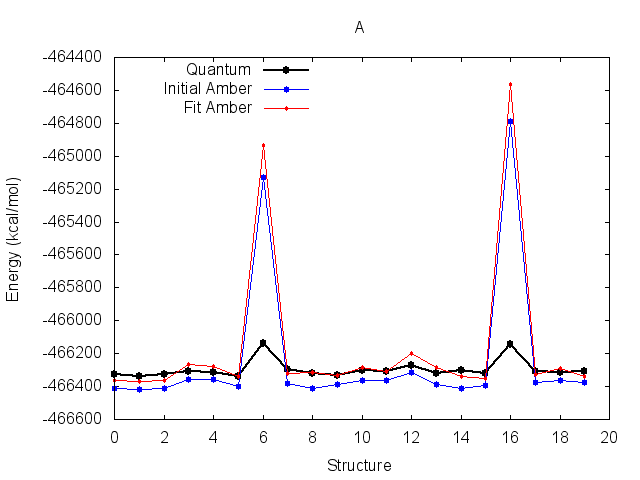
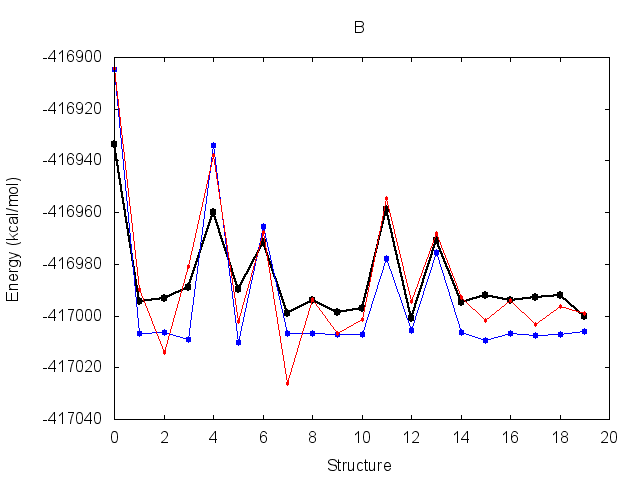
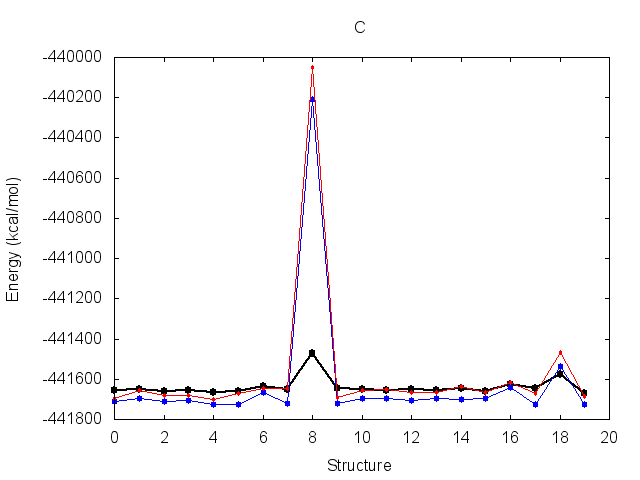
The output frcmod file can be used as input to leap when preparing new Amber topology files, using the loadamberparams <frcmod> command.
Conclusion
Congratulations! You have successfully completed the Paramfit tutorial. If you have any problems with Paramfit or this tutorial, first consult the Amber manual (section II.10), then email the Amber users mailing list.