


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
Derive K
When fitting to QM energies, Paramfit conducts a least squares fit to derive parameters. This means
meaning that it adjusts parameters so that the least squares difference between the input quantum
energy and calculated AMBER energy over all the input structures is minimized, using the following equation:
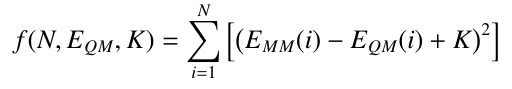 Where N is the input set of structures, EQM is the set of quantum energies of those structures,
EMM is the calculated AMBER energy of those structures, and K is a system-dependent constant
term.
Where N is the input set of structures, EQM is the set of quantum energies of those structures,
EMM is the calculated AMBER energy of those structures, and K is a system-dependent constant
term.
However, the origin is at a different location for these two sets of energies, which makes a perfect fit impossible. Since only the relative difference in energy between structures is important, we normalize the two sets of energies by adding a constant term, K, to the least squares fit. This term represents the difference between origins.
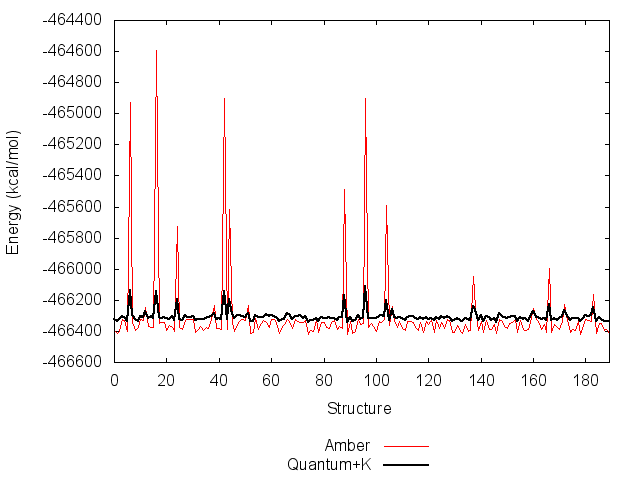
The value of K depends on the molecule and set of input structures, and is obtained by fitting using Paramfit.
Run Paramfit without any options to enter the job control wizard. Follow the prompts to conduct a fit for K only, using the AMBER standard energy fitting function. The resulting job control file will look something like this (without comments):
fit_K_A.in
# Use the first 20 structures of the input trajectory NSTRUCTURES=20 COORDINATE_FORMAT=TRAJECTORY # We want to fit the K parameter RUNTYPE=FIT PARAMETERS_TO_FIT=K_ONLY # Fit to quantum energies that are in units of Hartree FUNC_TO_FIT=SUM_SQUARES_AMBER_STANDARD QM_ENERGY_UNITS=HARTREE
Now invoke Paramfit with the topology, coordinate file, job control file, and the quantum input file created in the previous step:
paramfit -i fit_K_A.in -p a.prmtop -c A_valid_structures.mdcrd \
-q quantum_A.dat > fit_K_A.out
Output: (Yours will differ)
fit_K_A.out
Paramfit will give a lot of output here, but should finish nearly instantly. Look under FINAL PARAMETERS for the value of K for this system, and write it down.
------------------------------- FINAL PARAMETERS ---------------------------------
Parameters for force field equation: AMBER_STANDARD:
(* means parameter is NOT constant during fit)
*K = -466524.473754 kcal/mol
(CT-HC) Kr = 340.0000 kcal/(mol A)^2, r_eq = 1.0900 A
Also take note of the line indicating the final function value and R2:
Function value with fitted parameters = 3717020.1590, R^2 = 0.9440Remember, the algorithm tries to fit to zero- this seems to be really bad! Don't forget that we are just fitting K for now though.
Next, we will define what parameters to fit.