


Generating Force Field Parameters with Paramfit
Formerly known as Parmfit Tutorial
By Robin M. Betz
TABLE OF CONTENTS
IntroductionSystem setup
Generating conformations
Conduct quantum calculations
Derive K
Define parameters to fit
Visualise structure quality
Conduct single molecule fit
Weight individual structures
Multiple molecule fits
Weighting structures
Paramfit has the capability of weighting structures differently in the fit, so that those with especially high or low energy do not excessively bias the results. By default, all structures have a weight of 1. Higher weights mean that the structure is more important, and lower less. A weight of 0 means that a structure will be ignored during the fit.
With each structure given a weight wi, the fitting function becomes:
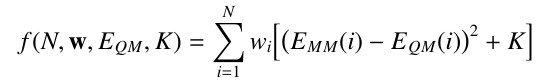
Assign weights
Weights are assigned in the second column of the file containing quantum energies. Previously, we only had the quantum energies in this file, resulting in the default weight of 1 being assigned to each structure. Now, make a copy of this file and add a second column to structures 6 and 16. Remember that the structures are numbered from 0, so this is actually line 7 and 17 in the energy file. The line should be readily identifiable by its much higher (less negative) energy. Assign these structures a weight of 0.0, meaning they will be ignored by the fit, as when we visualized the conformations they were completely unreasonable. All other structures will retain their default weight, so the column may be left blank for all other lines.
quantum_A_weighted.dat
Download
-743.137434317 -743.154591575 -743.137388686 -743.105020322 -743.117382187 -743.157255408 -742.834615002 0.0 -743.093303285 -743.129453943 -743.148561291 -743.100112704 -743.108246042 -743.050214136 -743.124268718 -743.103621696 -743.125943029 -742.843706235 0.0 -743.113225122 -743.123125683 -743.109442153
Re-derive K
Since the weight of each structure has changed, we need to re-derive K for this system. The same job control file for the previous derivation (fit_K_A.in) may be used when invoking Paramfit, but the quantum energy file with weights should be provided:
paramfit -i fit_K_A.in -p a.prmtop -c A_valid_structures.mdcrd \
-q quantum_A_weighted.dat > fit_K_A_weighted.out
Output: (Yours will differ)
fit_K_A_weighted.out
Check the top of the output file to ensure that weights were set for the correct structures:
Reading energy file or directory : quantum_A_weighted.dat Structure 6 weight = 0.000000 Structure 16 weight = 0.000000We get a significantly different value of K now, -466382.135709 (vs -466524.473749) kcal/mol.
Conduct weighted fit
We can use the same job control file as for the previous fit, but make sure to change the value of K to the new value, and save the output energy file to fit_output_energy_weighted.dat. Also set SEARCH_SPACE=-1 since we found the initial search area isn't a problem and we want maximal sampling of parameter space.
Run Paramfit (it may take a few minutes):
Download input
paramfit -i job_fit_weighted.in -p a.prmtop -c A_valid_structures.mdcrd \
-q quantum_A_weighted.dat > fit_weighted.out
Output: (Yours will differ)
fit_weighted.out
fit_output_energy_weighted.dat
frcmod
Watching the output, the initial fitness evaluation is much smaller than in the unweighted run. This is because the high energy structures are no longer included, and they had such a huge difference between quantum and Amber energy that they were the main contributor to the fitness function sum.
The resulting parameters from this fit are much more reasonable. Visualize the resulting energy file with the plot script, there is much less difference between quantum and fit Amber at the default scale.
However, the fit parameters result in a greater deviation between AMBER and quantum energy for the two structures that we ignored. This means that the resulting parameters do a worse job of describing the structure when it is in that conformation. For this run this is okay, as those conformations may reasonably be assumed to be energetically inaccessible during simulation.
When conducting your own fits though, be extremely careful to ensure that you are not ignoring reasonable structures, as weighting out structures just to obtain a "better" fit can result in physically meaningless parameters!
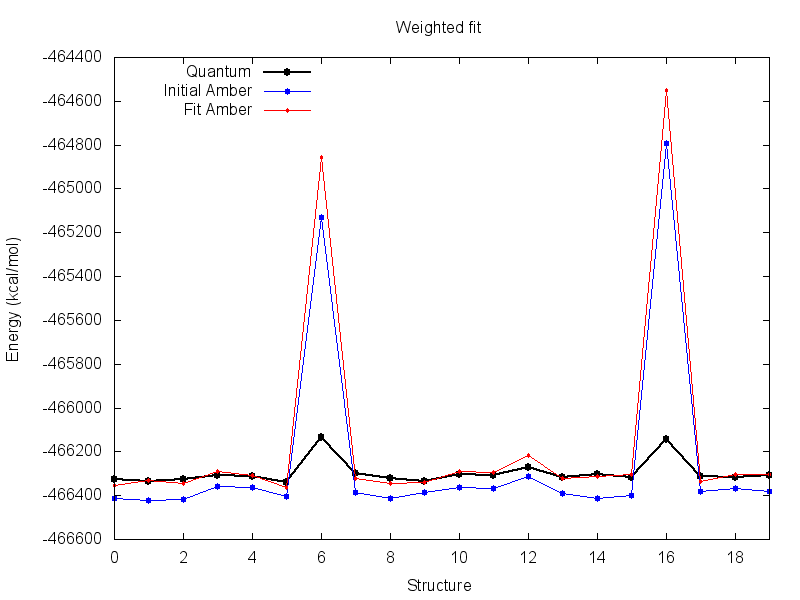
NOTE: It is a good idea to inspect the energy profile resulting from any fit.